






Mic19耗竭會損害內質網-線粒體接觸和線粒體脂質代謝,并引發肝病
內質網(ER)與線粒體接觸對于調節脂質轉運、合成和代謝至關重要。然而,內質網-線粒體接觸的分子機制和生理功能仍不清楚。研究人員發現MICOS(線粒體接觸位點和嵴組織系統)復合物的一個關鍵亞基Mic19通過EMC2-SLC25A46-Mic19軸調控ER-線粒體接觸。Mic19肝臟特異性敲除(LKO)會導致小鼠肝細胞內質網與線粒體接觸減少、線粒體脂質代謝紊亂、線粒體嵴紊亂和線粒體未折疊蛋白應激反應,損害肝臟線粒體脂肪酸β氧化和脂質代謝,從而可能自發引起小鼠非酒精性脂肪性肝炎(NASH)和肝纖維化。然而,Mic19 LKO肝細胞中Mic19的重新表達可阻斷小鼠肝病的發展。此外,Mic19過表達還能抑制MCD誘導的脂肪肝。因此,作者的研究結果揭示了EMC2-SLC25A46-Mic19軸是調節ER-線粒體接觸的途徑,并揭示了ER-線粒體接觸受損可能是與NASH和肝纖維化發展相關的機制。本文于2024年1月發表在《Nature Communications》,IF:16.6。
技術路線
主要實驗結果:
1.Mic19參與調節ER-線粒體接觸
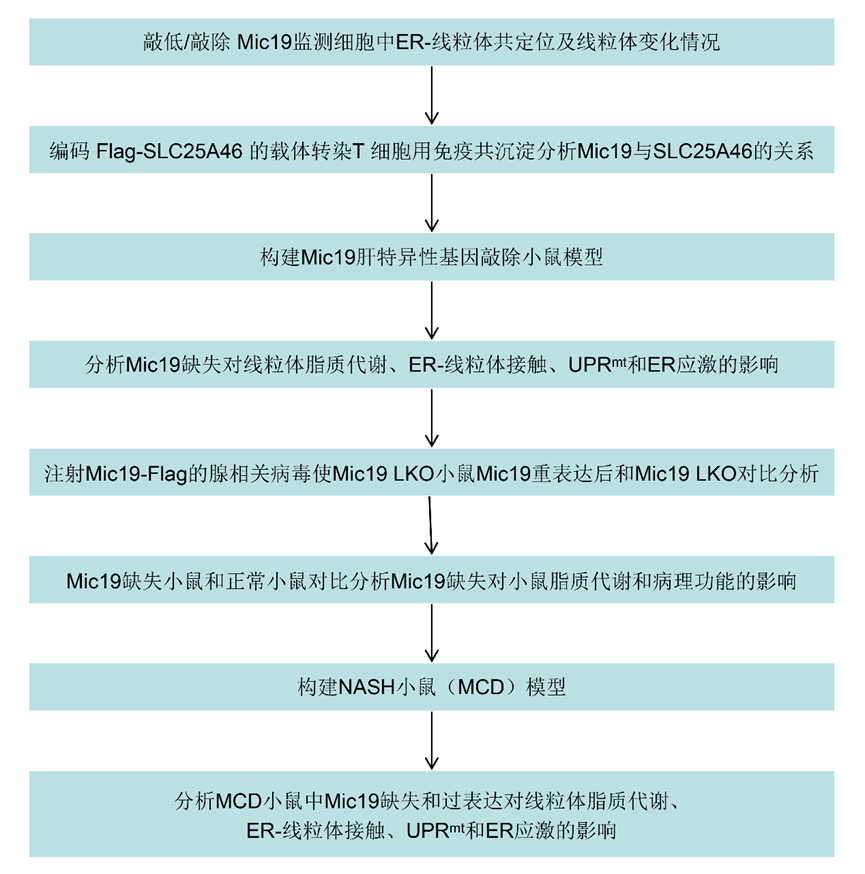
線粒體可與細胞內質網(ER)接觸,以調節重要的細胞穩態功能。MAMs是ER和線粒體之間的物理聯系,對細胞穩態至關重要。Monteiro-Cardoso等人最近報道,脂質轉移蛋白ORP5和ORP8主要位于MAM亞結構域,并與MICOS復合物物理連接。Mic19是MICOS復合物的一個關鍵亞基,對MICOS和SAM復合物的完整性至關重要。作者隨后研究了Mic19在線粒體-ER接觸中的作用。HIS-SIM分析顯示,在Mic19敲除(KD)或敲除(KO)的HeLa或COS7細胞中,ER和線粒體的共定位減少(圖1a-d)。此外,透射電子顯微鏡(TEM)分析表明,Mic19 KO導致線粒體超微結構異常,包括線粒體嵴連接(CJs)缺失和線粒體嵴排列改變,但線粒體長度沒有變化(圖1e-g)。此外,Mic19KO導致ER與線粒體之間的距離顯著增加,并導致ER-線粒體接觸數量減少(圖1h,i),表明Mic19耗竭會減少ER-線粒體接觸。此外,作者還研究了Mic19耗竭對線粒體數量和含量的影響,這可能會損害ER-線粒體接觸。HIS-SIM成像和Western blotting(WB)分析表明,線粒體數量和含量(線粒體標記蛋白,包括Tom20、Tom40、Tim23、SDHA和Cox4)在Mic19KO細胞中沒有變化。此外,mito-keima檢測顯示,與對照細胞相比,Mic19 KO細胞的線粒體自噬沒有發生變化。因此,這些結果表明,Mic19參與了ER-線粒體接觸的調控。
圖一:Mic19耗竭減少ER-線粒體接觸。
2.Mic19通過EMC2-SLC25A46-Mic19軸調節ER-線粒體接觸
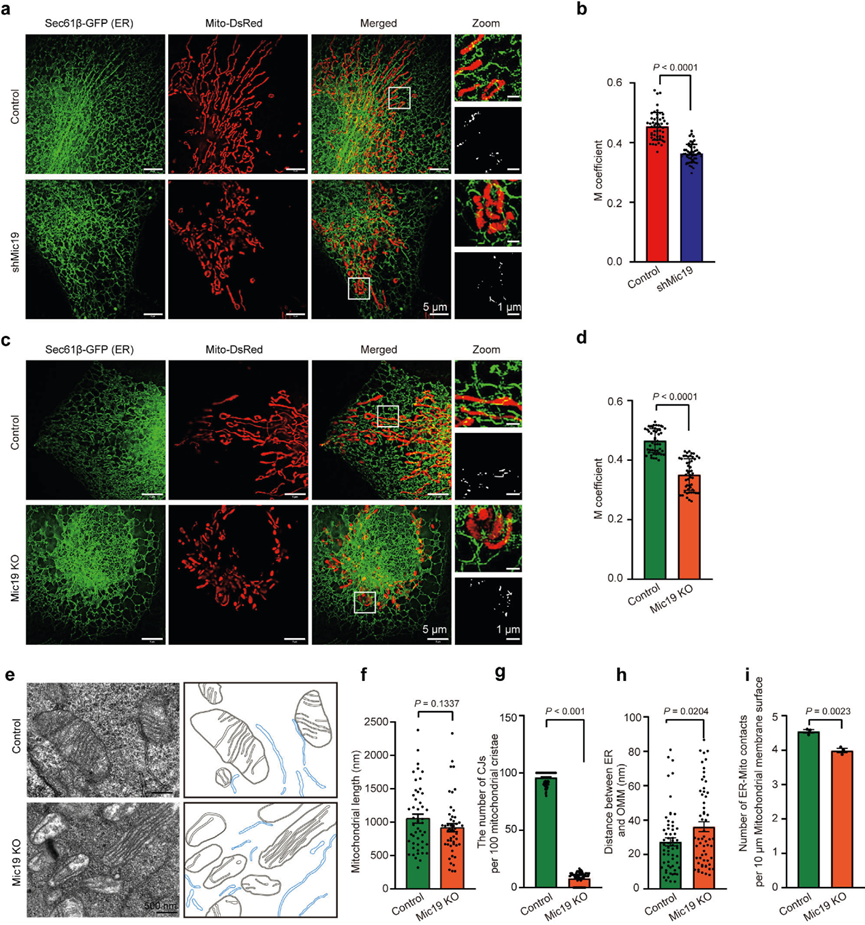
線粒體外膜蛋白SLC25A46與MICOS復合物相互作用,并通過EMC235維持與ER的相互作用。因此,作者研究了Mic19調控的ER與線粒體之間的串擾是否與SLC25A46有關。使用抗Flag或抗SLC25A46抗體進行免疫共沉淀(co-IP)分析表明,Flag-SLC25A46偶聯珠或SLC25A46偶聯珠能沉淀Mic19、Mic60和EMC2,而對照珠不能,表明SLC25A46能與Mic19、Mic60和EMC2相互作用(圖2a,b)。此外,Mic19 KO會降低細胞中SLC25A46的蛋白水平(圖2c,d),SLC25A46 KD也會導致Mic19蛋白的輕微減少(圖2e,f)。此外,MG132處理不能抑制CHX(環己亞胺,蛋白質合成抑制劑)誘導的對照組和Mic19 KO細胞中SLC25A46的減少(降解),表明Mic19KO引起的SLC25A46降解獨立于泛素-蛋白酶體途徑。然后,作者探討了線粒體蛋白酶對Mic19 KO誘導的SLC25A46降解的影響。WB分析顯示,OMA1或Yme1L的缺失會顯著抑制Mic19KO細胞中SLC25A46的降解,這表明線粒體蛋白酶OMA1和Yme1L有助于Mic19 KO引起的SLC25A46降解。此外,WB分析顯示,CLS1基因敲除降低了細胞中Mic19、Mic60和SLC25A46的蛋白水平,這表明SLC25A46和MICOS亞基的降解可能是心磷脂依賴性的。
此外,HIS-SIM成像顯示,SLC25A46 KD顯著減少了HeLa細胞中ER-線粒體的接觸(圖2g,h)。此外,TEM分析顯示,SLC25A46 KD導致HeLa細胞中CJs減少,但線粒體長度沒有改變(圖2i-k);而且,SLC25A46 KD顯著增加了ER和線粒體之間的距離,減少了線粒體-ER接觸的數量(圖2l,m),表明SLC25A46調控ER-線粒體接觸。值得注意的是,通過mito-keima檢測,SLC25A46 KD細胞的線粒體自噬沒有發生變化。因此,這些結果表明,Mic19與SLC25A46相互作用形成EMC2-SLC25A46-Mic19軸,有助于調控ER-線粒體接觸。
圖二:Mic19通過EMC2-SLC25A46-Mic19軸參與線粒體和ER之間的相互作用。
3.Mic19基因缺失誘導線粒體脂質代謝紊亂
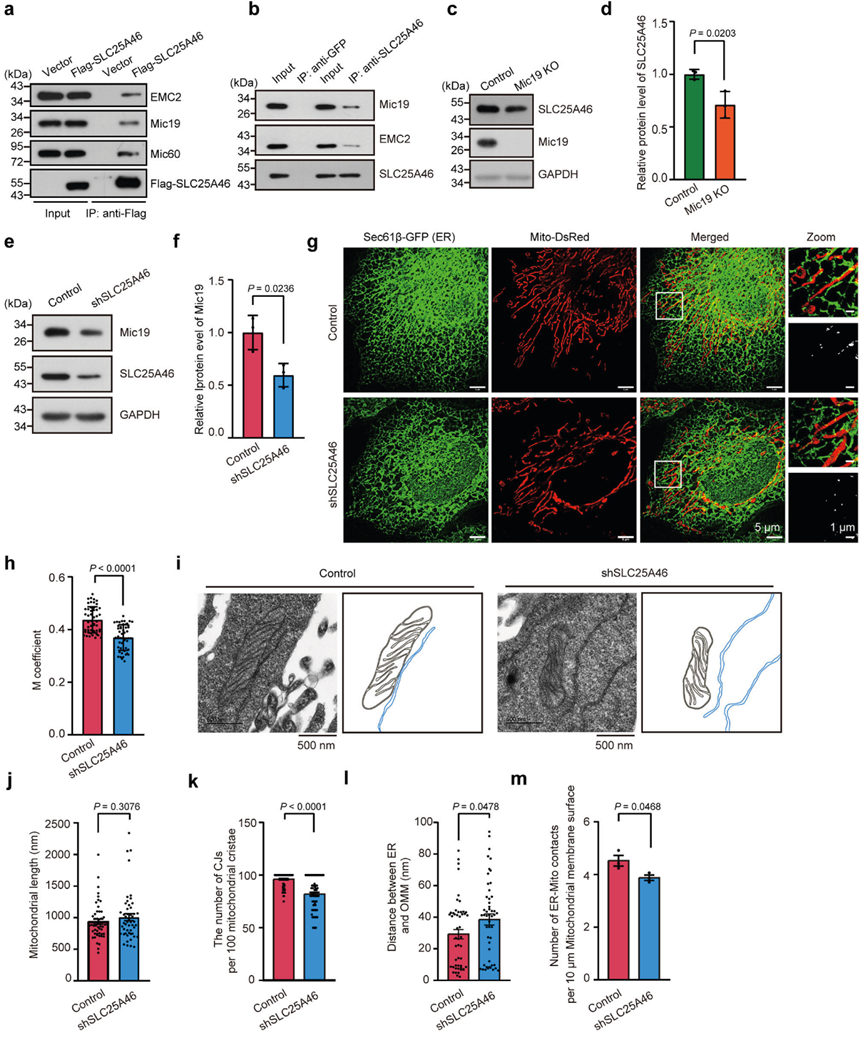
ER-線粒體接觸對于磷脂在ER和線粒體之間的運輸至關重要,可調節線粒體的脂質代謝。作者將Mic19flox/flox與在小鼠肝細胞中特異表達Cre的Albumin-Cre轉基因小鼠雜交,產生了Mic19肝特異性基因敲除(LKO)小鼠。WB分析表明,Mic19在小鼠肝臟中被特異性消耗,但在小鼠的其他器官,包括肌肉、心臟、脾臟、腎臟和大腦中也有表達。然后,作者分離了線粒體部分,分析了3個月大的Mic19 LKO小鼠肝臟中線粒體磷脂的含量。脂質組學分析表明,Mic19 LKO小鼠肝臟線粒體部分的磷脂酸(PA)、心磷脂(CL)、磷脂酰膽堿(PC)、磷脂酰絲氨酸(PS)和磷脂酰乙醇胺(PE)的豐度下降(圖3a,b),表明Mic19的缺失損害了線粒體磷脂的代謝。CL是線粒體特有的磷脂,主要位于IMM38,對線粒體結構和功能的維持至關重要。由于EMC2-SLC25A46-Mic19軸調控線粒體-ER接觸,作者研究了EMC2-SLC25A46-Mic19軸在CL代謝中的作用。作者分離了細胞的線粒體部分,并對純化的線粒體部分進行了WB分析。進一步的脂質組學分析表明,Mic19KO或SLC25A46 KD細胞線粒體部分的CL水平下降(圖3c-f)。此外,作者還通過10-N-壬基吖啶橙(NAO,一種心磷脂結合染料)染色分析了對照組、Mic19 KO和SLC25A46 KD細胞中的CL水平。共聚焦成像和流式細胞術分析表明,Mic19或SLC25A46的缺失導致CL顯著減少(圖3g-j),表明Mic19或SLC25A46的缺失會影響CL的代謝。此外,TMRM染色和流式細胞儀分析顯示,Mic19KO和SLC25A46 KD降低了細胞線粒體膜電位,這可能是由于CL代謝受損所致。此外,WB分析顯示,在Mic19 KO或SLC25A46 KD細胞中,CLS1(心磷脂合成酶1)和Tafazzin(TAZ,催化轉酰形成成熟的心磷脂)的蛋白水平沒有變化。此外,Mic19 LKO小鼠肝臟中的CLS1或TAZ蛋白水平也保持不變。這些數據表明,Mic19或SLC25A46的缺失并不影響線粒體合成CL的能力。值得注意的是,雖然CLS1和TAZ的蛋白水平沒有變化,但許多其他因素也可能導致心磷脂水平降低:磷脂合成酶的活性可能受到抑制,包括酶失活修飾;此外,底物限制也可能是一個因素。因此,Mic19基因缺失通過EMC2-SLC25A46-Mic19軸誘導線粒體脂質代謝紊亂。EMC2-SLC25A46-Mic19軸共同調節ER-線粒體接觸并參與線粒體脂質代謝。
圖三:線粒體和ER之間的異常串聯導致線粒體中的心磷脂減少。
4.Mic19基因敲除導致小鼠肝臟線粒體膜紊亂和線粒體未折疊蛋白反應
ER-線粒體接觸對于調控線粒體的多種功能至關重要。為了研究EMC2-SLC25A46-Mic19軸介導的ER-線粒體接觸的生物學和生理功能,作者培養了Mic19 LKO小鼠。Mic19 LKO導致包括Mic10、Mic60和Mic13在內的MICOS復合體亞基顯著減少,并導致SLC25A46顯著減少(圖4a,b)。由于Mic19的缺失會影響CL的產生(圖3g、h),而CL對線粒體膜的組織至關重要,并與氧化應激和線粒體功能障礙有關,因此作者評估了Mic19 LKO對小鼠肝臟線粒體膜超微結構的影響。TEM分析表明,與對照組(Mic19flox/flox小鼠肝細胞)相比,Mic19 LKO小鼠肝細胞顯示ER與線粒體之間的距離增加,ER-線粒體接觸數量減少(圖4c-e),證實Mic19 LKO損害了ER-線粒體接觸。此外,Mic19 LKO小鼠肝細胞線粒體嵴明顯減少,CJs急劇減少(圖4c,f,g),此外Mic19 LKO小鼠肝線粒體嵴膜急劇紊亂(圖4c)。因此,Mic19 LKO會導致小鼠肝臟線粒體膜重塑,尤其是線粒體嵴膜的紊亂。
線粒體嵴是線粒體氧化磷酸化的主要部位,而氧化磷酸化對細胞能量的產生至關重要。作者通過對從小鼠肝臟中分離出來的線粒體進行Blue Native-PAGE分析,評估了Mic19 LKO誘導的嵴解離對線粒體氧化磷酸化的影響。引人注目的是,在Mic19 LKO小鼠肝臟線粒體中,復合體I、復合體III、復合體IV和復合體V的水平顯著下降,而復合體II的水平卻沒有下降。接下來,作者利用Oroboros O2k系統的高分辨率呼吸測定法分析了3個月大的Mic19flox/flox和Mic19 LKO小鼠肝細胞的耗氧率。在各種線粒體復合物抑制劑的作用下,Mic19 LKO小鼠肝臟的耗氧量持續低于Mic19flox/flox小鼠肝臟的耗氧量(圖4h),表明Mic19 LKO導致小鼠肝臟線粒體氧化磷酸化缺陷。此外,Mic19 LKO小鼠肝臟顯示ATP生成減少和ROS水平升高,表明Mic19 LKO誘導的線粒體膜紊亂導致線粒體應激或功能障礙。線粒體應激或功能障礙期間,線粒體未折疊蛋白反應(UPRmt)被激活,導致包括線粒體蛋白酶和伴侶蛋白在內的保護基因轉錄上調。因此,作者研究了Mic19 LKO是否會觸發小鼠肝臟中的UPRmt。WB分析表明,在Mic19 LKO小鼠肝臟中,UPRmt相關蛋白包括線粒體蛋白酶(LONP1、ClpP)、線粒體伴侶HSP60和SOD2(UPRmt的替代標記物)的蛋白水平顯著升高,而Tom40(線粒體標記物)的蛋白水平沒有升高(圖4i、j),表明Mic19 LKO在體內導致了UPRmt。
此外,作者還評估了Mic19 LKO對小鼠肝臟ER穩態的影響。作者測試了Mic19 LKO對ER應激的影響。作者通過WB分析測定了多種標記物的表達,包括GRP78(ER應激整體傳感器)、Atf6(內質網穩態調節因子)、p-eIF2α(翻譯起始因子eIF2磷酸化)和Chop(ER應激下游蛋白)。與對照組相比,Mic19 LKO小鼠肝臟中的GRP78、Atf6、Chop和p-eIF2α蛋白水平顯著升高(圖4k、l)。此外,ER應激抑制劑TUDCA(tauroursodeoxycholate)能顯著抑制Mic19 LKO誘導的小鼠GRP78、Atf6、Chop和p-eIF2α的上調。這些結果表明,Mic19 LKO觸發了小鼠肝臟的ER應激。
綜上所述,Mic19 LKO引起的ER-線粒體接觸減少與UPRmt和ER應激高度相關。
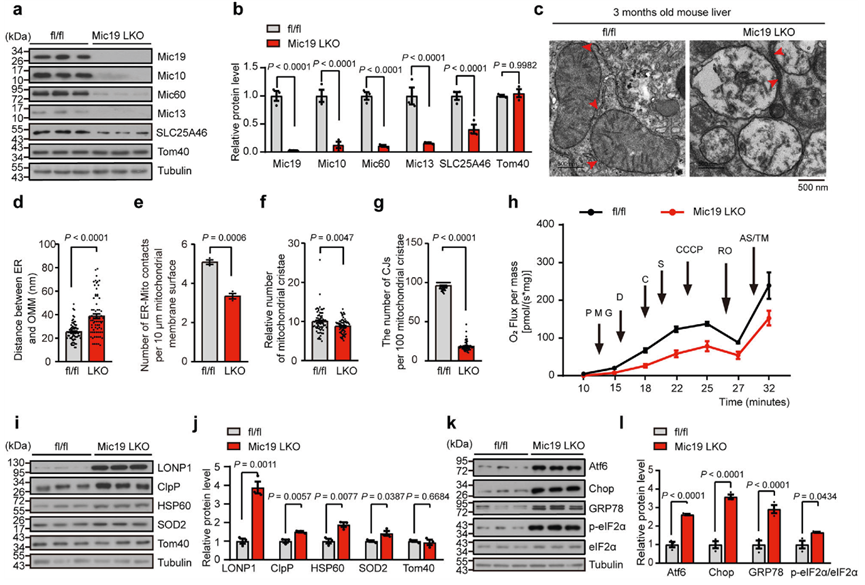
圖四:Mic19 LKO會導致小鼠出現UPRmt和ER應激。
5.Mic19 LKO會損害小鼠肝臟的脂肪酸代謝
為了進一步研究Mic19介導的ER-線粒體接觸的生理功能和線粒體脂質代謝紊亂,作者研究了Mic19 LKO對小鼠脂質代謝的影響。在正常飲食條件下,Mic19 LKO小鼠的體重比同窩對照小鼠(Mic19flox/flox)顯著下降,這并不是由于食物和水攝入量減少,因為Mic19 LKO小鼠的食物和水攝入量顯著增加。此外,Mic19 LKO小鼠3個月大時肝臟甘油三酯(TG)水平顯著升高,但肌肉甘油三酯、血清TG、肝臟膽固醇和血清極低密度脂蛋白(VLDL)水平沒有變化(圖5a-e)。此外,Mic19 LKO小鼠肝臟和血清游離脂肪酸(FFA)水平顯著升高(圖5f、g)。然而,Mic19 LKO小鼠肝臟中丙二酰-CoA水平和大多數脂肪生成相關基因(包括Mlycd、Fasn、Gpat2、Agpat1、Lpin1和Dgat2)的mRNA水平均無變化。此外,WB分析表明,在Mic19小鼠肝臟中,p-Acc1/Acc1(比值)和Fasn(負責從頭合成脂肪酸的主要酶)沒有發生變化,這表明Mic19 LKO可能不會影響脂肪酸的合成。這些數據表明,Mic19 LKO損傷了小鼠肝臟的脂肪酸代謝而非合成。為了進一步探索其潛在機制,作者提取了對照組和Mic19 LKO小鼠肝臟RNA進行測序。作者發現Mic19 LKO小鼠肝臟中有3145個基因上調,3133個基因下調。有趣的是,與脂質代謝相關的基因,尤其是與脂肪酸代謝相關的基因(細胞色素P450、亞油酸和花生四烯酸的代謝)在Mic19 LKO小鼠肝臟中被下調(圖5h)。因此,作者對小鼠肝臟的線粒體脂肪酸氧化進行了分析。qRT-PCR分析顯示,在禁食條件下,Mic19 LKO小鼠肝臟中一些線粒體β氧化基因包括Cpt1a、Cpt2、Acad9和Acads的mRNA水平顯著下降(圖5i)。然而,ER應激抑制劑TUDCA抑制了Mic19 LKO誘導的這些與線粒體β氧化有關的基因的減少。此外,在喂養和禁食條件下,Mic19 LKO小鼠肝臟中乙酰-coA的水平也明顯降低(圖5j)。這些數據表明,Mic19 LKO損傷了小鼠肝臟線粒體脂肪酸的β-氧化。
此外,眾所周知,線粒體脂肪酸β氧化可導致肝臟在禁食條件下產生酮體(生酮)。作者發現,在Mic19 LKO小鼠肝臟中,β-羥基丁酸(βHB,酮體之一)的生酮產物明顯減少(圖5k),生酮相關基因Hmgcs2和Hmgcl的mRNA水平下降(圖5l),表明Mic19 LKO小鼠肝臟的生酮過程受損。
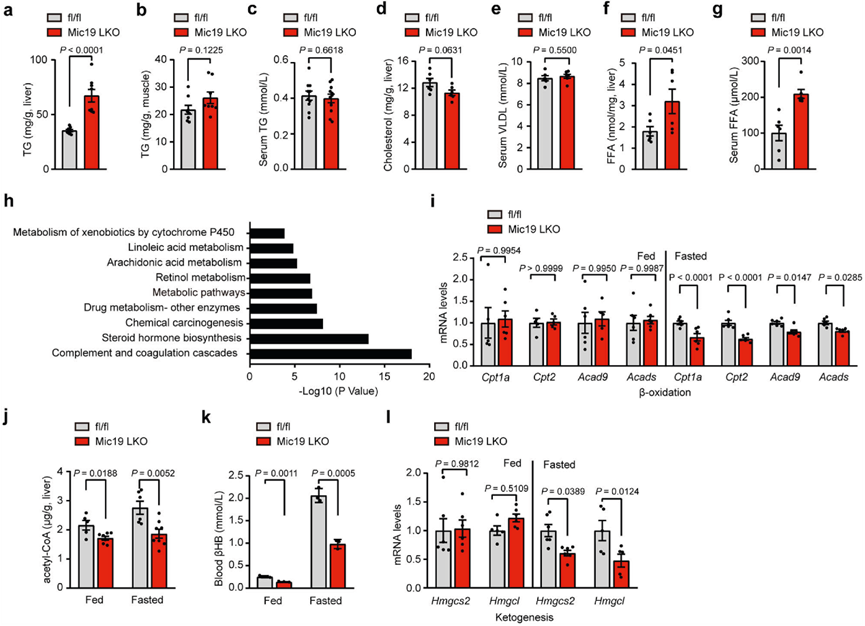
圖五:Mic19 LKO影響小鼠的脂肪酸代謝。
此外,在小鼠代謝籠實驗中,Mic19 LKO小鼠的耗氧量(VO2)和二氧化碳產生率(VCO2)也顯著增加。脂肪組織是調節能量平衡的重要器官,它不僅儲存能量,還是新陳代謝的調節器。包括eWAT(附睪白色脂肪組織)和iWAT(腹股溝白色脂肪組織)在內的脂肪組織重量略有下降。然而,qRT-PCR分析顯示,在Mic19 LKO小鼠iWAT中,一些線粒體β氧化基因(包括Cpt1a、Cpt2和Acads)的mRNA水平顯著升高。這些結果表明,Mic19 LKO小鼠iWAT中脂肪分解增加,這可能是肝組織中β氧化功能受損的代償反應,從而增加了Mic19小鼠的能量消耗。
因此,Mic19 LKO增加了小鼠肝臟中脂肪酸和總膽固醇的水平,這很可能是由于肝線粒體β氧化作用的減少。
6.Mic19 LKO會導致非酒精性脂肪性肝炎(NASH)和肝纖維化
由于Mic19 LKO會損害小鼠肝臟線粒體的β氧化功能,作者隨后評估了Mic19 LKO對小鼠病理功能的影響。與對照組相比,Mic19 LKO使小鼠3個月大時丙氨酸氨基轉移酶(ALT)和天門冬氨酸氨基轉移酶(AST)的活性顯著升高(圖6a、b),表明Mic19 LKO會導致小鼠肝損傷。此外,Mic19 LKO小鼠(3個月大)的肝臟顯示出大量脂肪堆積(圖6c)。此外,蘇木精和伊紅(H&E)染色以及油紅O染色顯示,Mic19 LKO小鼠(3個月大)的肝臟出現了明顯的脂肪變性(圖6d、e)。進一步的TEM分析顯示,與對照組小鼠相比,Mic19 LKO小鼠(3個月大)肝臟中脂滴(LDs)的數量和大小明顯增加(圖6f-h)。此外,在喂養或禁食條件下,Mic19 LKO小鼠肝臟(3月齡)的CD36(脂肪酸轉運體,脂肪肝的標志物)mRNA水平是對照組小鼠肝臟的10倍以上,表明脂質攝取可能是Mic19 LKO誘導小鼠肝臟脂質積累的原因之一。這些數據表明,Mic19 LKO會導致3個月大的小鼠出現脂肪肝。此外,作者還研究了Mic19 LKO誘導的脂肪積累是否會導致肝臟炎癥。作者檢測了編碼炎癥細胞因子和趨化因子的基因的表達。qRT-PCR分析顯示,在Mic19 LKO小鼠(3月齡)肝臟中,炎癥相關基因Cxcl10、Cd68和Tnf的mRNA水平顯著升高。免疫組化分析CD68染色進一步顯示Mic19 LKO小鼠(3個月大)肝臟中單核細胞/巨噬細胞明顯增加,證實Mic19 LKO會導致3個月大小鼠慢性肝臟炎癥。此外,Masson三色染色顯示,對照組和Mic19 LKO小鼠(3個月大)的肝臟沒有差異。因此,作者的數據表明,Mic19 LKO會導致小鼠(3個月大)發生非酒精性脂肪性肝炎(NASH)。
慢性肝臟炎癥可導致肝纖維化。作者接下來研究了Mic19 LKO是否會導致肝纖維化,這是肝病自然進展的結果。在正常飲食條件下,Mic19 LKO小鼠(7個月大)的體重仍然低于同窩對照組。此外,7個月大的Mic19 LKO小鼠血清中的谷丙轉氨酶(ALT)和谷草轉氨酶(AST)仍然顯著升高(圖6i、j)。然而,與對照組相比,Mic19 LKO小鼠在7個月大時并未出現明顯的脂肪堆積(圖6k)。此外,對照組和Mic19 LKO小鼠(7個月大)的肝臟TG水平沒有差異(圖6l),這可能是由于小鼠的代償效應。H&E染色顯示Mic19 LKO小鼠(7個月大)肝臟中炎癥和壞死區域明顯增加(圖6m),qRT-PCR分析顯示Mic19 LKO小鼠(7個月大)肝臟中炎癥相關基因包括Cxcl10、Cd68和Tnf的mRNA水平明顯增加。此外,Masson三色染色顯示,與對照組相比,Mic19 LKO(7個月)導致小鼠肝臟細胞外膠原蛋白急劇積累(圖7n),表明Mic19 LKO導致小鼠(7個月)肝纖維化。此外,qRT-PCR分析表明,在Mic19 LKO小鼠(7個月大)中,編碼纖維化標志物(包括Col1a1和Col3a1)的基因的mRNA水平顯著升高。此外,Mic19 LKO小鼠(7個月大)體內的羥脯氨酸(肝組織中的纖維化標志物)、血清堿性磷酸酶(ALP,肝病標志物)和γ-谷氨酰轉肽酶(γ-GT,肝細胞損傷標志物)水平也顯著升高。這些數據表明,Mic19 LKO會導致肝纖維化逐漸加重。總之,Mic19 LKO會逐漸引發小鼠的NASH和肝纖維化。
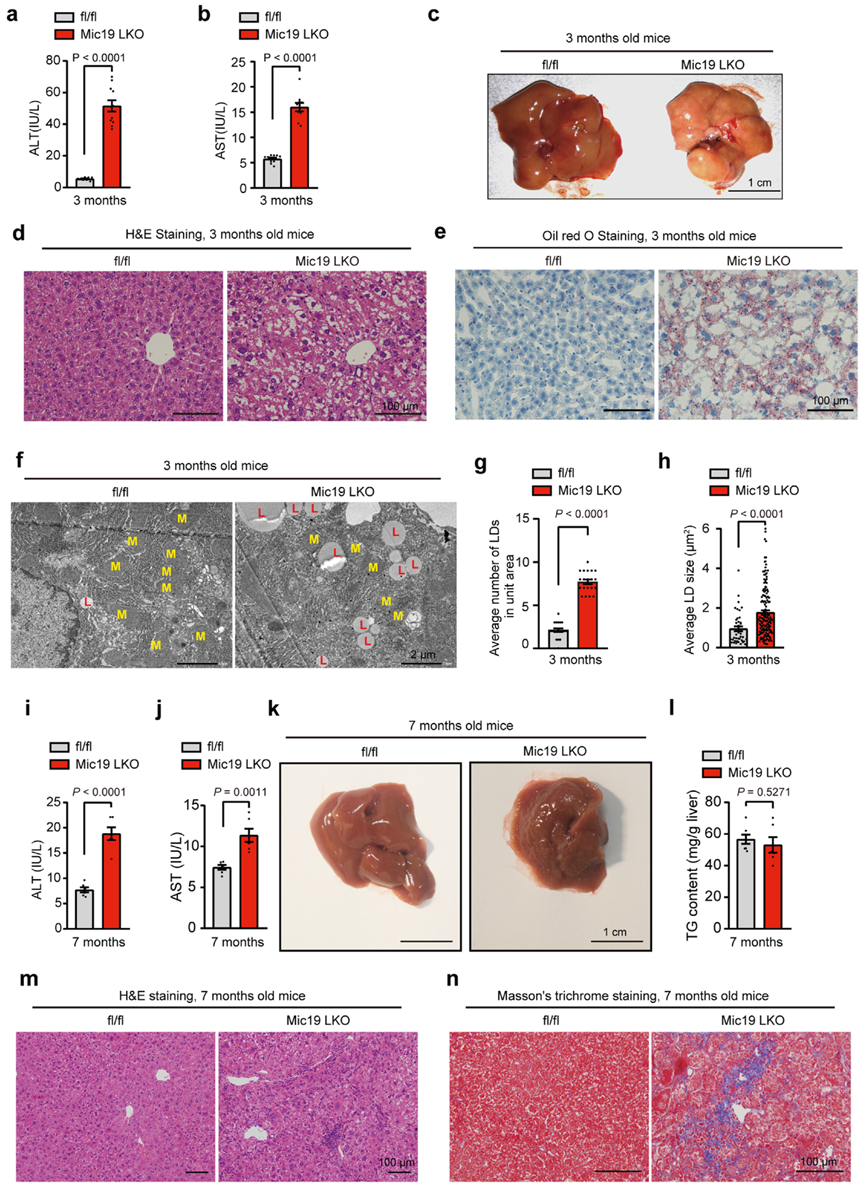
圖六:Mic19 LKO導致小鼠患上非酒精性脂肪肝。
7.Mic19 LKO小鼠體內重新表達Mic19可恢復肝臟脂質代謝并阻止肝臟疾病的發生
為了確定Mic19 LKO小鼠的改變是否歸因于Mic19功能缺失機制,作者給8周齡的Mic19 LKO小鼠尾部靜脈注射編碼對照組或Mic19-Flag的腺相關病毒(AAV),使Mic19在小鼠肝臟中重新表達。在Mic19 LKO小鼠肝臟中重新表達Mic19后(圖7a),小鼠的體重恢復到對照組(Mic19flox/flox)小鼠的體重,UPRmt相關蛋白包括LONP1、ClpP、HSP60和SOD2的蛋白水平明顯低于Mic19 LKO小鼠肝臟的蛋白水平,與對照組小鼠肝臟的蛋白水平沒有差異(圖7b,c),表明Mic19的再表達極大地抑制了Mic19 LKO誘導的UPRmt。此外,作者還研究了Mic19重表達對小鼠ER應激的影響。WB顯示,重表達Mic19 LKO小鼠肝臟中的GRP78、Atf6、Chop和p-eIF2α蛋白水平顯著低于Mic19 LKO小鼠肝臟中的水平,但與對照組小鼠肝臟中的水平相似,表明重表達Mic19顯著減輕了Mic19 LKO引起的小鼠ER應激。
然后,作者研究了Mic19重表達對小鼠肝臟脂質代謝的影響。在Mic19 LKO小鼠肝臟中重新表達Mic19后,其肝甘油三酯(TG)水平明顯低于Mic19 LKO小鼠(圖7d)。同樣,油紅O染色顯示,Mic19的再表達極大地抑制了Mic19 LKO引起的肝脂肪變性(圖7e)。此外,與Mic19 LKO相比,Mic19的再表達顯著下調了肝臟炎癥相關基因(包括Cxcl10、Cd68和Tnf)的mRNA水平(圖7f)。此外,Mic19的再表達還顯著降低了小鼠血清中谷丙轉氨酶(ALT)和谷草轉氨酶(AST)的水平(圖7g、h)。這些數據表明,重新表達Mic19可通過恢復肝臟脂質代謝阻斷Mic19 LKO誘發的NASH。
作者還研究了重新表達Mic19是否能抑制Mic19 LKO引起的小鼠(7個月大)肝纖維化。H&E和Masson的三色染色顯示,Mic19的再表達顯著減少了Mic19 LKO(7個月)導致的小鼠肝臟壞死和細胞外膠原蛋白的積累(圖7i-l)。此外,qRT-PCR分析表明,Mic19重表達Mic19 LKO小鼠(7個月大)肝臟中編碼纖維化標志物Col1a1和Col3a1的基因的mRNA水平明顯低于Mic19 LKO小鼠肝臟中的水平(圖7m)。這些數據表明,Mic19的再表達可阻斷Mic19 LKO(7個月)誘發的小鼠肝纖維化。因此,在Mic19 LKO小鼠中重新表達Mic19可以通過恢復肝臟脂質代謝來阻止Mic19 LKO引發的NASH和肝纖維化。
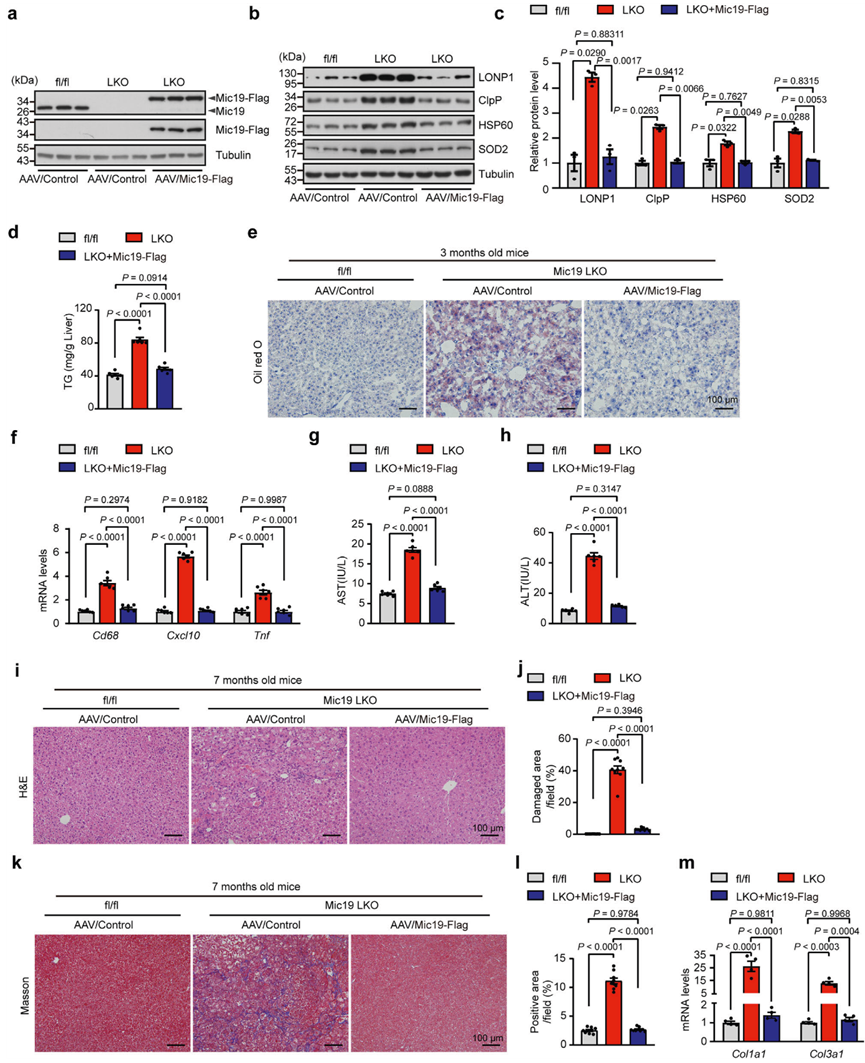
圖七:重新表達Mic19可改善Mic19 LKO小鼠的肝損傷。
8.Mic19過表達抑制MCD誘導的脂肪肝
為了進一步證實Mic19與NASH之間的聯系,作者建立了NASH小鼠模型(稱為MCD),該模型由蛋氨酸和膽堿缺乏飲食與45%高脂飲食(HFD)組成,并在飲用水中補充0.1%的L-蛋氨酸。H&E和油紅O染色顯示,MCD小鼠的脂肪堆積明顯增加(圖8a、b)。此外,MCD小鼠肝臟中的總膽固醇水平和炎癥相關基因Cd68、Cxcl10和Tnf的mRNA水平也明顯升高(圖8c,d)。這些數據表明,MCD會導致小鼠的NASH(脂肪肝和肝臟炎癥)。然后,作者檢測了Mic19在對照組和MCD小鼠中的表達。MCD小鼠肝臟中Mic19和Mic60蛋白的表達量明顯低于對照組(圖8e,f)。此外,TEM分析顯示,MCD小鼠肝細胞中線粒體嵴明顯減少,線粒體嵴連接點數量急劇下降(圖8g-i),表明MCD誘導線粒體嵴重塑,這與MCD小鼠肝臟中Mic19蛋白水平較低的數據一致。有趣的是,與對照組相比,MCD小鼠肝細胞中的ER線粒體接觸明顯減少(圖8g、j)。
然后,作者研究了過表達Mic19對MCD誘導的肝病的影響。將編碼Mic19-Flag的腺相關病毒經尾靜脈注射到8周齡的MCD小鼠體內。MCD小鼠肝臟過表達Mic19-Flag后,TG水平和炎癥相關基因Cd68、Cxcl10和Tnf的mRNA水平的上調均被抑制,并恢復到與正常飲食小鼠相似的水平(圖8k-m)。此外,H&E染色顯示,Mic19-Flag的過表達顯著抑制了MCD引起的小鼠肝臟脂肪變性(圖8n)。因此,Mic19的過表達抑制了MCD誘導的脂肪肝。這些結果與Mic19抑制小鼠肝病的功能一致。
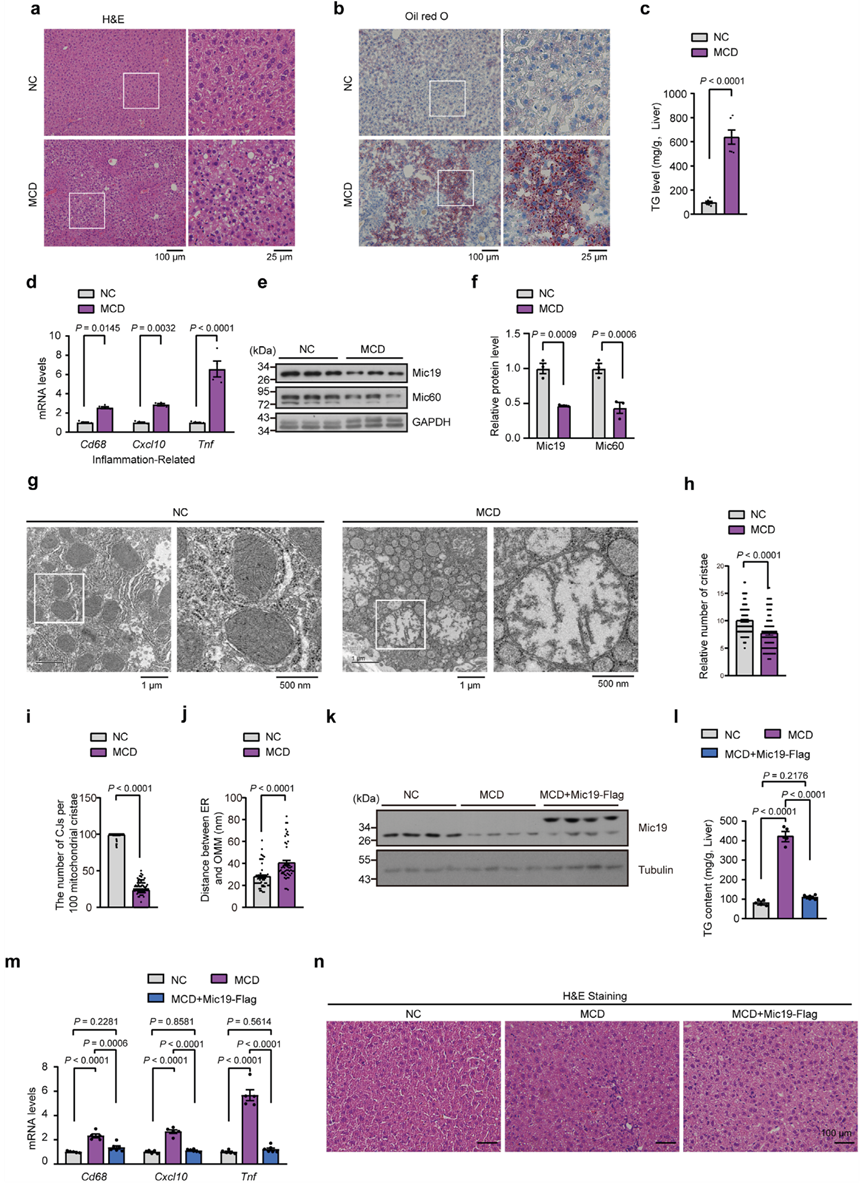
圖八:患有MCD誘導的脂肪肝的小鼠模型顯示肝臟的Mic19水平較低。
實驗方法:
蛋白免疫印跡(WB)、免疫共沉淀(co-IP)、免疫熒光(IF)、免疫染色和共聚焦成像、透射電子顯微鏡(TEM)、流式細胞術、qRT-PCR、免疫組化分析
參考文獻:
Dong J, Chen L, Ye F, Tang J, Liu B, Lin J, Zhou PH, Lu B, Wu M, Lu JH, He JJ, Engelender S, Meng Q, Song Z, He H. Mic19 depletion impairs endoplasmic reticulum-mitochondrial contacts and mitochondrial lipid metabolism and triggers liver disease. Nat Commun. 2024 Jan 2;15(1):168. doi: 10.1038/s41467-023-44057-6. PMID: 38168065; PMCID: PMC10762189.