






NRF2通過與TOPBP1協同激活ATR-CHK1信號通路促進輻射抵抗
肺癌是一種致命的惡性腫瘤,每天死亡人數超過350人。臨床研究表明約77%的肺癌患者接受放療(Radiotherapy,RT)。盡管放療的療效顯著改善,但癌細胞在連續暴露于電離輻射(Ionizing Radiation,IR)時產生抗性導致疾病復發。IR主要通過引起DNA損傷,特別是雙鏈斷裂(DSBs)來殺死癌細胞。輻射抗性癌細胞已經發展出強大的DNA損傷修復能力來生存IR。核因子?紅細胞2相關因子2(Nuclear Factor Erythroid 2-related Factor 2,NRF2)與輻射抗性相關。在本研究中,作者采用TCGA數據庫和組織芯片分析NRF2與肺癌患者預后的相關性,構建耐輻射肺癌細胞,通過體內和體外實驗探討NRF2在抗輻射中的作用,并采用免疫沉淀、免疫熒光和染色質級分提取等方法探討其潛在機制。本研究證明NRF2通過與RPA32和TOPBP1合作激活ATR-CHK1信號通路增強輻射抗性,還驗證了NRF2可作為放療的潛在靶點。
該研究于2024年1月1日發表在《Theranostics》,IF:12.4
技術路線
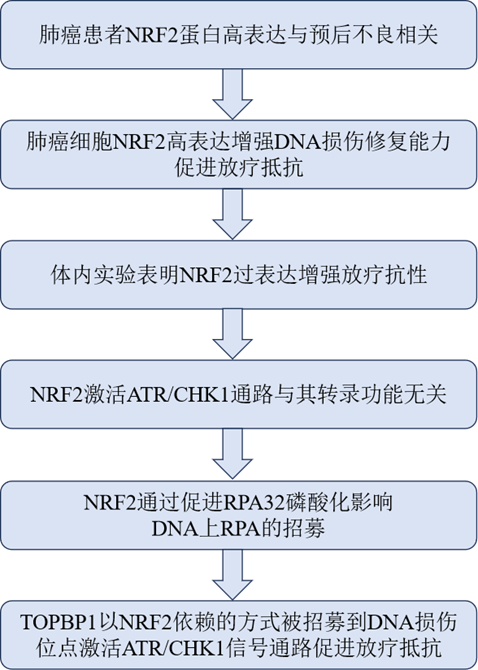
主要研究結果
1.NRF2/KEAP1基因改變引起NRF2激活與肺癌患者預后不良相關
作者通過c-BioPortal網站分析了肺癌患者NRF2/KEAP1基因的變異類型。在分析了來自29項研究的12,478個樣本/ 9,930名肺癌患者后,發現NRF2和KEAP1基因的總變異分別為4%和13%(圖1A)。遺傳突變是NRF2和KEAP1基因變異的主要形式。在拷貝數變異類型中,擴增是NRF2的主要改變類型,而KEAP1的主要改變類型是缺失(圖1A)。肺癌的不同亞型,如肺腺癌(LUAD)和肺鱗狀細胞癌(LUSC),具有不同的細胞變異類型,導致不同的生長特征。此外,作者發現NRF2的擴增和突變主要發生在LUSC(圖1B),KEAP1基因變異在LUAD中發生更頻繁(圖1C)。接下來通過RNA-seq分析表明,LUAD和LUSC中NRF2的主要CNV類型是擴增和增益,這導致NRF2 mRNA表達增加(圖1D-E)。KEAP1的主要變異類型是淺缺失,伴隨著較低的KEAP1 mRNA表達(圖1F-G)。此外還調查了NRF2/KEAP1基因改變組和NRF2/KEAP1基因未改變組患者之間的臨床結果。結果表明,NRF2/KEAP1基因的基因改變與較短的總生存期相關(圖1H-I)。NRF2/KEAP1基因改變患者的總生存期中位數分別為38.24和38.14個月,而無基因改變組分別為56.35和54.34個月。因此,上述結果表明,NRF2/KEAP1基因變異導致NRF2表達水平升高,并與肺癌患者預后不良有關。
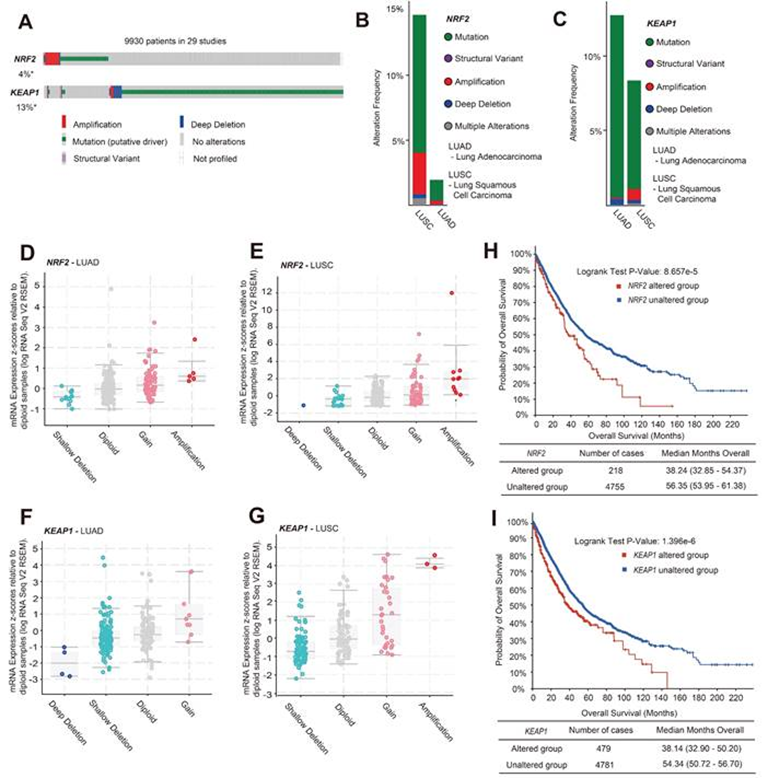
圖1:NRF2/KEAP1變異導致的NRF2過表達與肺癌患者預后不良相關
IHC檢測由LUAD患者和鄰近非癌組織作為對照的肺癌組織樣本組成的腫瘤微陣列(TMA)中NRF2蛋白的表達。用NRF2抗體染色TMA,并根據染色強度評估評分。根據細胞質中NRF2染色強度將LUAD樣本分為高/中/低NRF2表達水平的三組(圖2A)。在LUAD組中,44%的患者屬于NRF2高表達組(圖2B),該組患者的中位壽命為31.23個月(圖2C)。接下來作者根據NRF2蛋白在細胞核中的定位將患者分為陽性和陰性組,結果顯示核NRF2檢測陽性的患者的壽命明顯短于檢測陰性的患者(圖2D-E)。
總體而言,TMA結果與TCGA數據庫分析結果一致,表明肺癌患者NRF2蛋白高表達與預后不良相關。
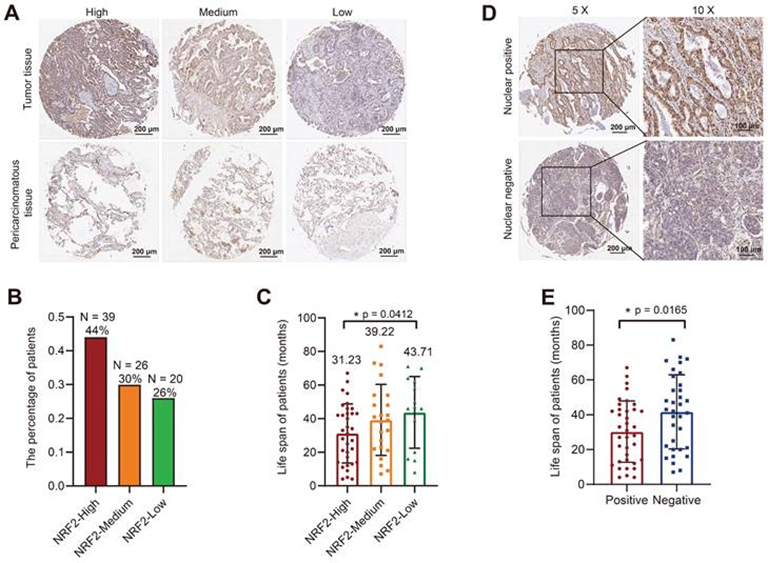
圖2:組織芯片染色NRF2與LUAD患者預后的相關性分析。
2.放療抵抗細胞中NRF2高表達
作者建立了人類肺癌抗輻射細胞并通過集落形成實驗進行驗證。對肺癌細胞和肺癌放療抵抗細胞使用質譜進行蛋白質組學分析,發現氧化應激信號通路發生變化。檢測了輻射抵抗細胞中NRF2的蛋白水平,發現與A549細胞相比,A549R細胞中具有抗氧化應激功能的NRF2蛋白及其下游血紅素氧合酶-1(HO-1)顯著上調(圖3A)。NRF2向細胞核轉移是激活下游基因的先決條件,這些基因編碼一系列II期解毒或抗氧化酶。核質分離結果顯示,與H460和H1299細胞相比,H460R和H1299R細胞核中NRF2蛋白水平顯著升高(圖3B-C)。此外,免疫熒光實驗顯示,在H1299R細胞中NRF2熒光信號更強烈(圖3E)。這些結果表明NRF2在輻射抗性細胞中上調,并優先積累在細胞核中。
有研究報道過NRF2可通過激活ATR/CHK1通路導致DSBs和G2/M細胞周期停滯。因此作者接下來監測了放射抗性細胞中ATR信號級聯的變化。WB結果顯示,IR誘導H460和H1299細胞中ATR和CHK1的磷酸化,H460R和H1299R細胞中ATR和CHK1的磷酸化水平明顯更高(圖3F-G)。采用免疫熒光監測γ-H2AX以評估DNA損傷。紅外線照射后,A549R細胞γ-H2AX病灶的數量顯著低于A549細胞,表明輻射抵抗細胞的DNA修復能力優于對照細胞(圖3H)。H460和H1299細胞也觀察到類似的趨勢(圖3I-J)。上述結果表明,輻射抵抗細胞中高水平的NRF2蛋白增強了DNA損傷修復能力,這可能是肺癌細胞可以發生輻射抵抗的原因之一。.
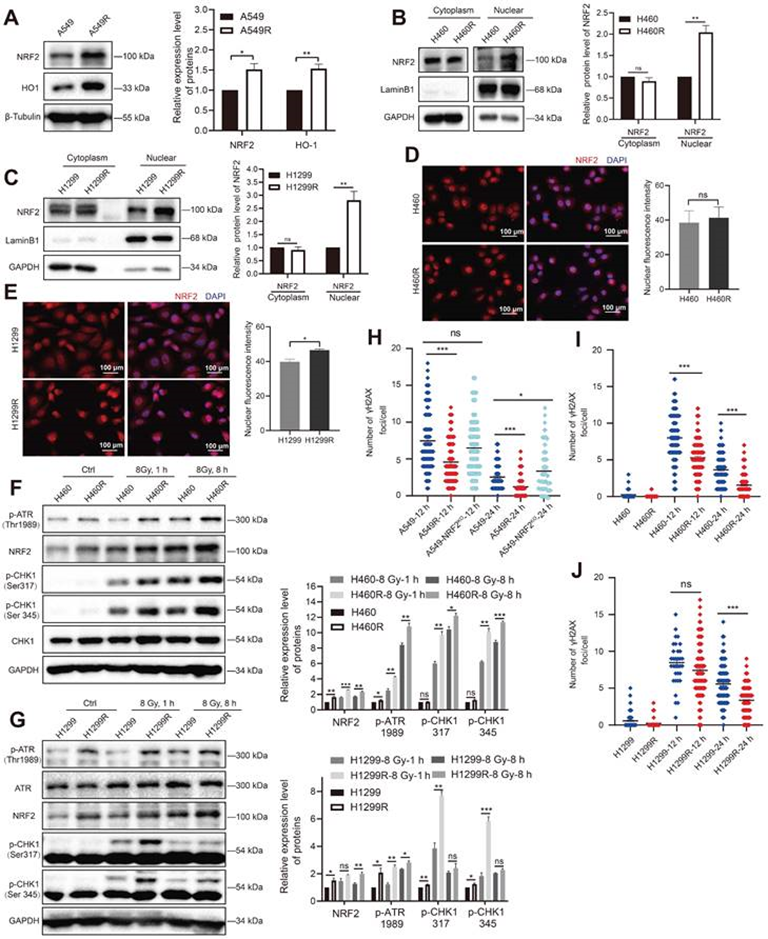
圖3:NRF2在抗輻射細胞中表達顯著
3. 體內NRF2過表達的抗輻射細胞對放療的抗性更強
將穩定表達GFP的A549和A549R細胞注射到裸鼠尾靜脈,6周后成功構建肺腫瘤轉移模型,將小鼠分為對照組(A549)、抗輻射細胞組(A549R)、對照照射組(A549-IR)、抗輻射細胞照射組(A549R-IR)四組。每周用2或4 Gy分次劑量的γ射線局部照射肺部,治療持續3周(圖4A)。熒光信號檢測到肺部腫瘤轉移(圖4B)。與A549組相比,A549-IR組放療后肺腫瘤轉移數量明顯減少。相比之下,在注射A549R細胞的小鼠中,接受或不接受放療的肺轉移數量沒有觀察到統計學上的顯著差異,提示A549R腫瘤對IR更具抗性(圖4C-D)。結果顯示A549R細胞在體內也具有抗輻射性。肺組織切片IHC分析顯示,A549細胞源性腫瘤中的Ki67陽性染色在照射后顯著下降,但A549R-IR組與A549R-未照射組的Ki67豐度差異較小,提示IR可能不能有效抑制A549R細胞的增殖(圖4E)。此外,用IHC檢測NRF2的染色強度。A549組的NRF2染色在照射后顯著下降,而A549R組的變化不顯著(圖4F)。總的來說,這些研究結果表明NRF2蛋白與體內肺癌細胞的輻射抵抗之間存在關聯。
接下來,作者通過Nrf2+/ -C57BL/6小鼠雜交得到Nrf2+/+和Nrf2-/-幼鼠,以進一步探索NRF2在體內響應IR中的作用(圖4G)。將Nrf2+/+和Nrf2-/-小鼠暴露于兩種劑量的IR(7.2和6.5 Gy)(圖4H),發現Nrf2-/-小鼠表現出更高更快的死亡率(圖4I和4K)。Nrf2-/-小鼠的平均體重在全身照射(TBI)后繼續下降;而,Nrf2+/+小鼠的平均體重在TBI后大約20天趨于穩定(圖4J和4L)。這些結果表明,缺失NRF2基因導致輻射暴露后小鼠死亡率較高,這意味著NRF2可能在體內對輻射反應中發揮保護作用。
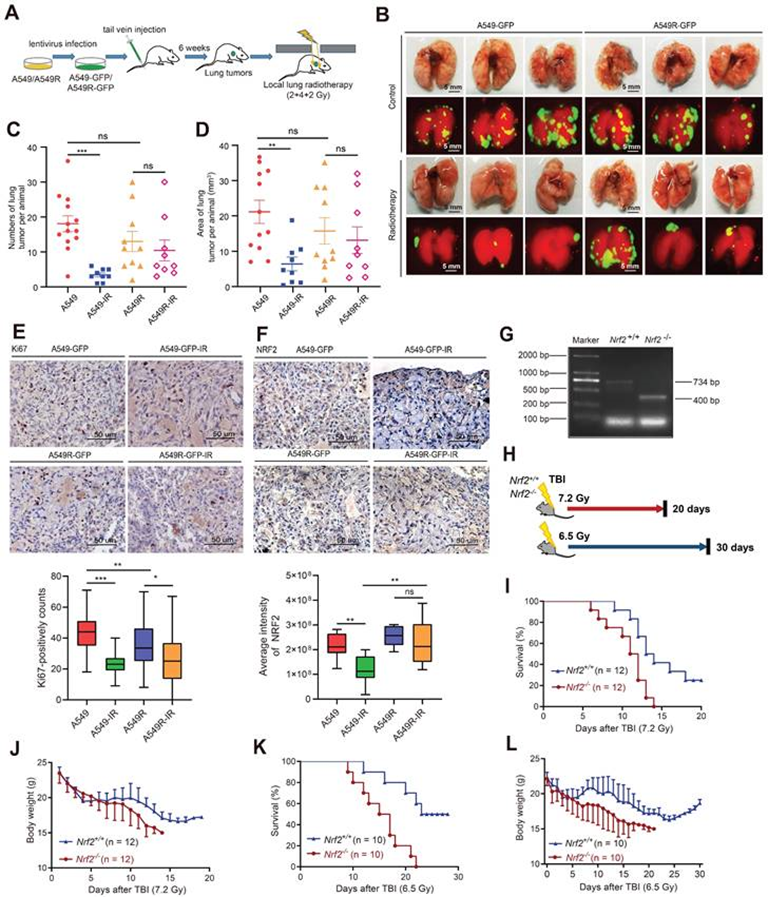
圖4:NRF2過表達的放射抗性細胞在體內對放療的抗性更強
4. NRF2激活ATR/CHK1通路獨立于其轉錄功能
接下來,作者試圖闡明NRF2導致放射抗藥性的機制。在A549和H1299細胞中,照射以劑量依賴性的方式增加NRF2蛋白水平(圖5A-B);8 Gy照射以時間依賴性的方式增加NRF2蛋白水平(圖5C-D)。之前的實驗結果表明NRF2蛋白在肺癌組織細胞核中的積累與肺癌預后較差有關(圖2D-E),抗輻射細胞細胞核中的NRF2蛋白水平也較高(圖3B-C)。因此,從細胞中分離出染色質,并檢查照射后NRF2與染色質的結合。結果表明,照射后NRF2蛋白與染色質的結合顯著增加(圖5E),這表明NRF2在紅外下轉移到細胞核并與染色質結合。此外,通過免疫熒光研究NRF2的亞細胞定位。結果表明,NRF2被招募到激光微照射引起的DNA損傷位點,并在H1703和A549細胞中與γ-H2AX共定位(圖5F)。
NRF2作為一種轉錄因子在應激條件下調節BRCA1和53BP1的表達。BRCA1和53BP1都是DNA損傷反應過程中的關鍵蛋白。在A549細胞中敲低NRF2后53BP1和BRCA1蛋白的水平顯著下降;而53BP1和BRCA1的敲低并不影響CPT處理后ATR和CHK1的激活(圖5G)。這些結果表明53BP1和BRCA1可能不是NRF2/ATR/CHK1信號通路激活的介導者。為了確認NRF2促進ATR-CHK1信號通路的激活不依賴于其轉錄調節功能,作者構建了刪除Neh1結構域的NRF2突變質粒(Δ434-561-NRF2)(圖5H),并在A549-NRF2 KO細胞中分別表達HA-NRF2和Δ434-561-NRF2。當細胞用8 Gy IR處理時,HA-NRF2和Δ434-561-NRF2的表達都顯著促進ATR和CHK1磷酸化(圖5I)。這些結果表明NRF2可以保持獨立于轉錄功能的基因組穩定性。
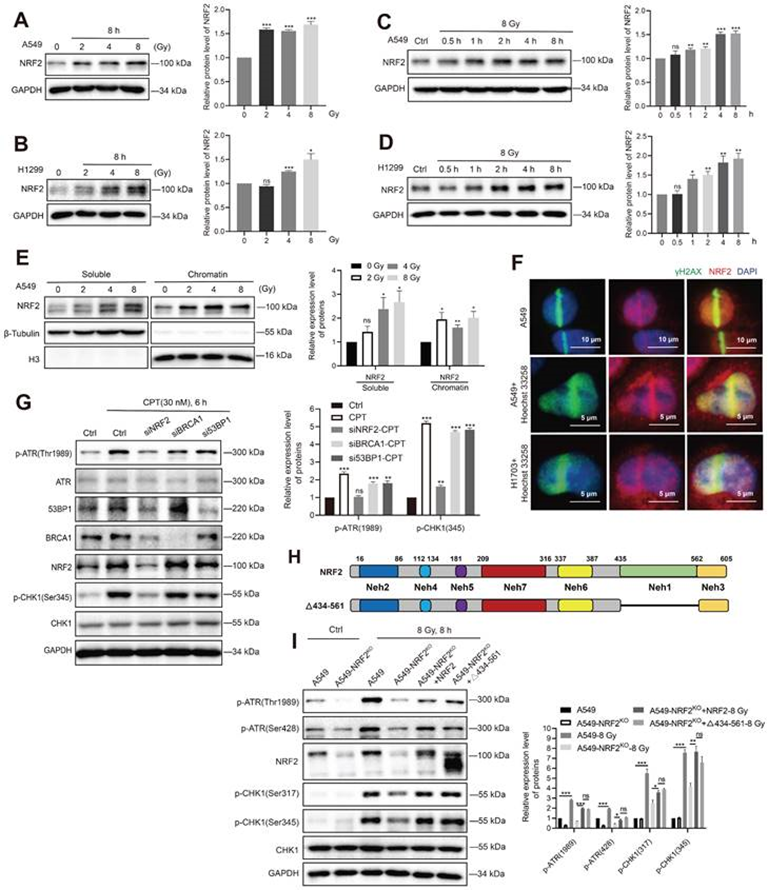
圖5:NRF2激活ATR/CHK1通路獨立于其轉錄功能
5. NRF2促進RPA32磷酸化和RPA在ssDNA的積累
NRF2可以直接結合并激活ATR,而RPA調節ATR激活。因此,作者探討了NRF2是否可以影響RPA在DNA損傷過程中的作用。RPA70和RPA32是RPA復合物的兩個主要亞基。在照射后6、12和24小時檢查了用siNRF2處理或未處理的A549細胞中RPA32和RPA70病灶的數量。免疫熒光結果顯示,敲低NRF2后A549細胞中RPA70病灶陽性細胞的比例明顯高于A549細胞暴露于IR后(圖6A)。同樣,敲低NRF2降低了細胞暴露于IR后RPA32病灶陽性細胞的比例(圖6B-C)。DNA結合的RPA與許多蛋白質相互作用以調節DNA代謝,RPA封裝的ssDNA也是復制應激誘導的DNA損傷反應過程的平臺。接下來,提取染色質結合蛋白以確定NRF2是否影響RPA32蛋白與DNA的結合,發現NRF2的敲除在照射后顯著抑制RPA32蛋白與染色質的結合,但不影響RPA32的可溶性形式(圖6D)。結果暗示NRF2可能有助于RPA32與染色質的結合在DNA損傷條件下形成病灶。
進一步研究了NRF2是否會影響RPA32的磷酸化。在A549細胞中,NRF2的敲低降低了CPT處理后RPA在Ser33和Thr21的磷酸化(圖6E)。此外還觀察到RPA32(Ser33、Thr21和S4/S8)的磷酸化水平在照射后10小時內持續增加,且A549R細胞中RPA32的磷酸化增加更加明顯(圖6F)。NRF2敲除細胞中RPA32的磷酸化明顯低于野生型細胞,并且通過在CPT(圖6G)處理后將Flag-NRF2重新引入A549-NRF2KO細胞來挽救這一缺陷。因此,研究結果表明NRF2通過促進DNA損傷過程中RPA32的磷酸化來影響DNA上RPA的招募。
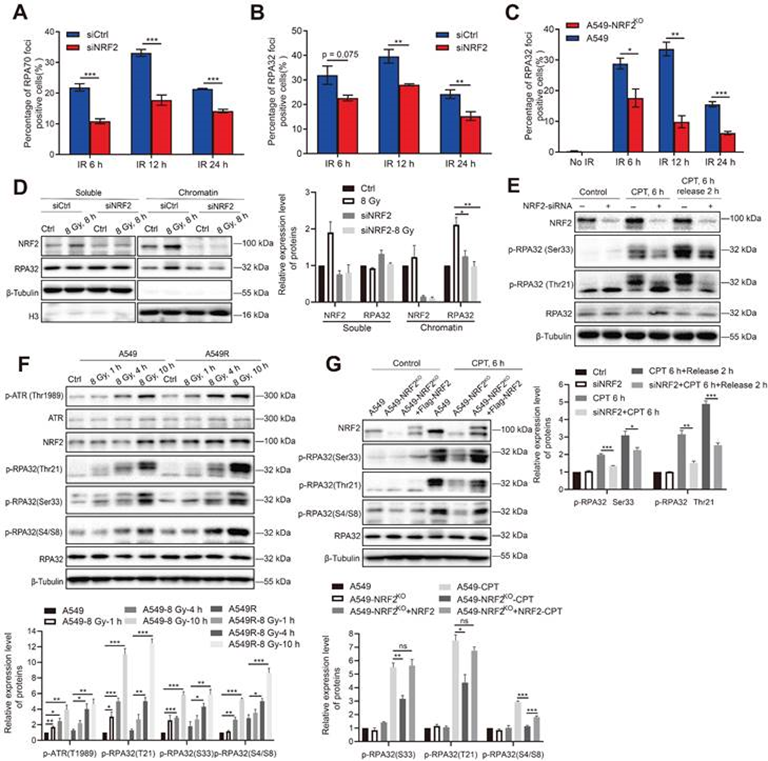
圖6:NRF2促進RPA32的磷酸化和RPA在ssDNA的積累。
6. NRF2協同TOPBP1激活ATR/CHK1信號通路
RPA32和CHK1都是ATR的磷酸化底物。有研究表明,TOPBP1和ETAA1的激活對應于不同的ATR功能。ETAA1在正常和應激復制過程中負責ATR介導的RPA磷酸化,而TOPBP1激活ATR/CHK1信號通路以響應復制應激。NRF2、TOPBP1和ETAA1在A549細胞中分別敲除,以比較NRF2、TOPBP1和ETAA1之間的差異。結果表明,轉染siNRF2的A549細胞在8Gy照射后ATR和CHK1的磷酸化水平比轉染siTOPBP1和siETAA1的細胞更顯著降低,表明NRF2在ATR的磷酸化中發揮了重要作用(圖7A)。而與單獨敲除NRF2相比,同時敲除TOPBP1和NRF2并沒有進一步降低ATR磷酸化水平(圖7A)。這些觀察結果表明NRF2可能與TOPBP1在同一途徑中發揮作用。因此,構建的Flag-TOPBP1和HA-NRF2融合蛋白進行驗證,用60 nM CPT處理細胞后,siTOPBP1在H1299R細胞中ATR/CHK1沒有磷酸化(圖7B)。表達HA-NRF2沒有挽救這一缺陷,這表明TOPBP1在NRF2/ATR/CHK1途徑中的關鍵作用(圖7B)。同時,Flag-TOPBP1的表達也未能磷酸化ATR和CHK1。
TOPBP1通過與RAD9-HUS1-RAD1(9-1-1)的復合物被招募到DNA損傷位點,并通過其AAD樣區域結合介導ATR的磷酸化。在染色質結合蛋白檢測實驗中,WB顯示NRF2的下調顯著抑制了TOPBP1蛋白與輻照A549細胞染色質的結合(圖7D)。使用靶向HPRT基因的Cas9/sgRNA實現了位置特異性DNA雙鏈斷裂,以進一步證實NRF2在TOPBP1招募到DNA損傷位點中的作用。熒光顯微鏡顯示TOPBP1與A549細胞中的γH2AX共定位。NRF2缺乏顯著減弱了A549-NRF2KO細胞中TOPBP1病灶的強度(圖7E-F)。圖7G結果清楚地表明,A549細胞中激光微輻射后TOPBP1被招募到DNA損傷位點,但在A549-NRF2KO細胞中沒有。這些結果表明,NRF2可以影響DNA損傷期間TOPBP1招募。接下來使用Co-IP驗證DNA損傷中TOPBP1和NRF2之間的相互作用。從A549細胞的染色質和可溶性提取物中免疫沉淀NRF2后可檢測到TOPBP1。此外,在IR后細胞的染色體提取物中TOPBP1和NRF2之間的相互作用增強(圖7H)。這些結果表明NRF2與TOPBP1合作促進ATR/CHK1信號通路的激活,并對照射后TOPBP1向DNA損傷位點的募集有促進作用。
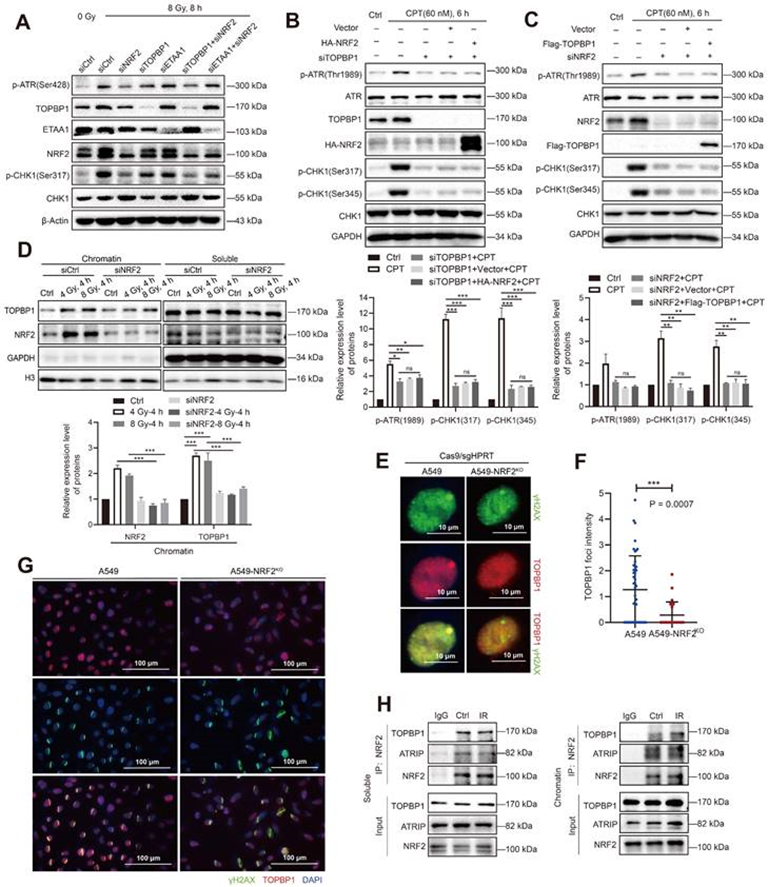
圖7:NRF2協同TOPBP1激活ATR/CHK1信號通路
結論
作者證明NRF2的高核表達與肺癌患者的不良預后相關,并有助于輻射抗性的發展。響應IR或CPT,NRF2被轉移到DNA損傷位點,促進RPA32的磷酸化,并通過招募TOPBP1到DNA損傷位點激活ATR/CHK1通路(圖8)。這項研究表明NRF2可能是改善肺癌放療的一個有希望的靶點。
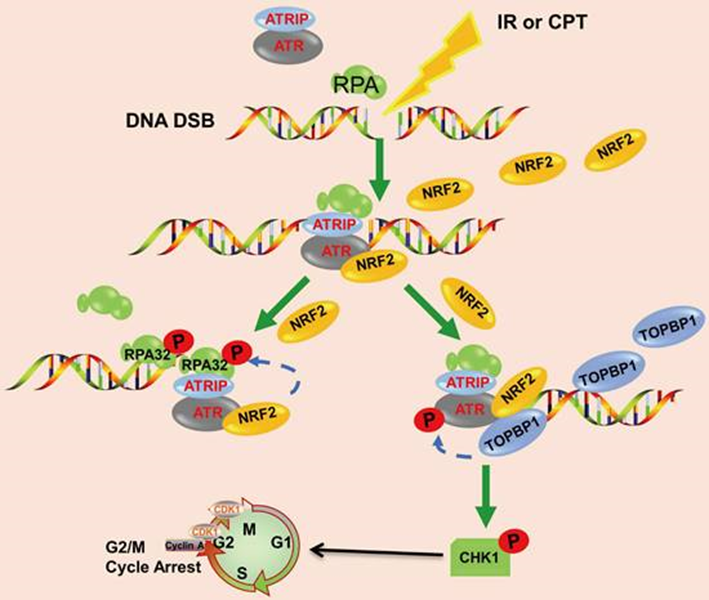
圖8
實驗方法
細胞培養和藥物治療,組織微陣列,輻射抗性細胞系構建,全細胞裂解物收集和蛋白質印跡法,免疫熒光分析,Co-IP實驗,活性氧水平的測定,集落形成試驗,腫瘤球形成實驗,核和細胞質蛋白質提取,提取染色質部分,慢病毒轉染實驗,siRNA轉染實驗,質粒轉染實驗,小鼠實驗,免疫組織化學染色,TUNEL實驗
參考文獻
Sun Xiaohui, Dong Mingxin, Li Jiale, et al. NRF2 promotes radiation resistance by cooperating with TOPBP1 to activate the ATR-CHK1 signaling pathway. [J]. Theranostics, 2024, 14: 681-698.