






METTL1介導的tRNA m7G修飾通過tRNA調控的翻譯控制促進AML的白血病生成
RNA修飾已被證實在調節細胞生物學過程中發揮著重要作用。最近,N7-甲基鳥苷(m7G)修飾及其修飾因子METTL1/WDR4已被證實在多種癌癥中具有致癌基作用。本文旨在確定METTL1/WDR4在急性髓系白血病(AML)中的功能和分子機制,更好地了解AML的白血病發生機制,為AML治療提供了有希望的候選靶點,提高AML的臨床治愈率。通過qRT-PCR、急性髓細胞性白血病臨床樣本的免疫印跡分析和公開急性髓細胞性白血病數據集的生物信息學分析,對METTL1/WDR4的表達水平進行量化。采用CCK-8檢測和細胞計數檢測確定細胞增殖情況。使用流式細胞儀檢測評估細胞周期和凋亡率。體外試驗機制研究采用了多種技術,如Northern印跡分析、液相色譜-串聯質譜(LC-MS/MS)、tRNA穩定性分析、轉錄組測序、小非編碼RNA測序、定量蛋白質組學和蛋白質合成測量。結果表明METTL1和WDR4在AML患者中顯著上調,其表達水平與AML患者預后不良密切相關。且敲除METTL1可有效抑制AML細胞增殖,促進細胞凋亡,并在G1期阻止細胞周期,從而抑制體外AML細胞進展。METTL1可通過特異性介導tRNA上m7G修飾狀態,調節細胞內攜帶m7G修飾tRNA的穩定性和生物發生,影響胞內涉及翻譯調節過程和酶活性等相關蛋白翻譯效率。該研究證實了METTL1介導的m7G-tRNA修飾在AML進展中的重要作用,為未來有效AML治療策略的開發提供了理論基礎。本文于2024年1月發表在《Experimental Hematology & Oncology》,IF:10.9。
技術路線
主要實驗結果:
1. METTL1和WDR4在AML中上調,與預后不良相關
為了評估METTL1/WDR4在AML中的表達,從NCBI GEO數據庫中收集了涉及健康個體和新診斷AML患者的公開數據集。骨髓細胞轉錄組分析顯示,與健康個體相比,AML患者METTL1和WDR4的表達水平顯著升高(圖1A)。新橋醫院對新診斷的AML患者和健康供體的公開數據集和qRT-PCR驗證的進一步比較分析表明,METTL1和WDR4在骨髓和外周血樣本中均顯著上調(圖1B、C)。在亞型分析中,METTL1和WDR4在幾乎所有FAB亞型中都表現出上調,尤其是在M1、M2、M4、M5和M6亞型中(圖1D、E)的AML。此外,根據第五次WHO分類標準重新分析了具有易位和突變亞型的AML患者中METTL1/WDR4復合物的豐度。在具有完整臨床數據的樣本中,與健康供體相比,大多數WHO亞型中METTL1和WDR4的表達上調。此外還分析了公共數據集以證實在患者中的觀察結果,結果與實驗數據相似。Western blot分析也證實,METTL1和WDR4在AML患者的骨髓單核細胞中表達更高(圖1F)。此外,使用來自癌癥基因組圖譜(TCGA)的急性髓系白血病(LAML)數據集發現METTL1或WDR4表達水平顯著較高的AML患者生存率較差(圖1G),這表明METTL1和WDR4在調節AML進展中具有潛在功能。
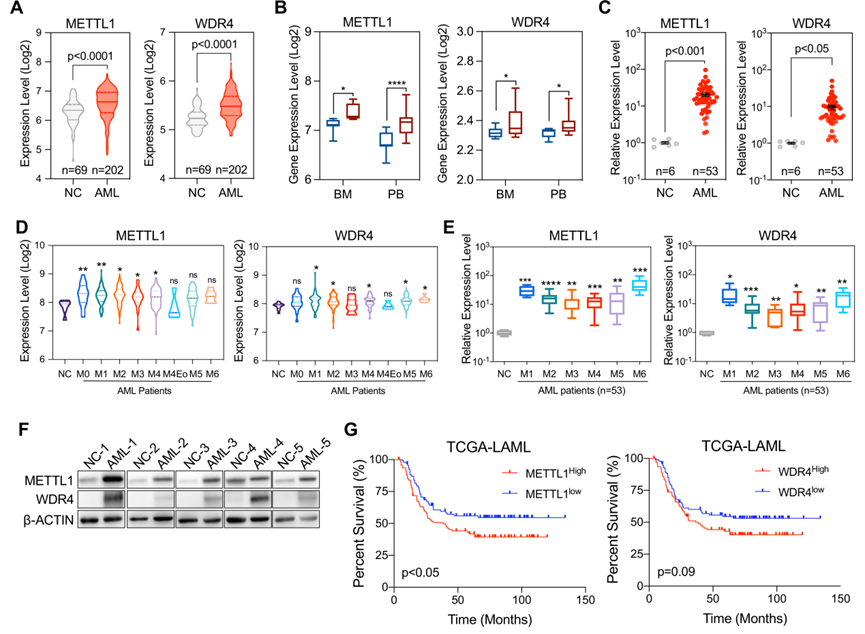
圖一:METTL1/WDR4上調與AML患者預后不良有關
2.METTL1敲除可抑制人AML細胞的存活和生長
為了研究METTL1在AML中的作用,我們使用shRNA系統來敲除METTL1的表達水平,其中包括一個陰性對照(shNC)和兩個獨立的shMETTL1(shMETTL1-1:shM1-1和shMETTL1-2:shM1-2),它們在THP-1和MOLM-13 AML細胞系中顯示出有效的METTL1蛋白水平敲除(圖2A)。結果顯示敲除METTL1抑制了上述AML細胞的細胞生長速率(圖2B、C)。細胞周期分析顯示,在AML細胞中敲除METTL1后,G1期細胞顯著增加,S期細胞減少(圖2D、E),表明敲除METTL1誘導細胞周期停滯在G1期。此外,膜聯蛋白V/7-AAD染色顯示,敲除METTL1誘導AML細胞凋亡(圖2F-H)。蛋白質印跡分析顯示,METTL1敲除導致BCL2下調,促進Caspase3的裂解,從而提高了AML細胞的凋亡率(圖2I)。相反,METTL1在HL60AML細胞中的過表達促進了細胞增殖(圖2J,K)。當接受阿糖胞苷(CYT)治療時,METTL1敲除的AML細胞表現出顯著降低的增殖能力和對阿糖胞苷誘導的細胞死亡的易感性增加(圖2L、M)。相反,用阿糖胞苷處理的過表達HL60細胞的METTL1表現出顯著加速的生長速率和減少的細胞凋亡(圖2N-P)。
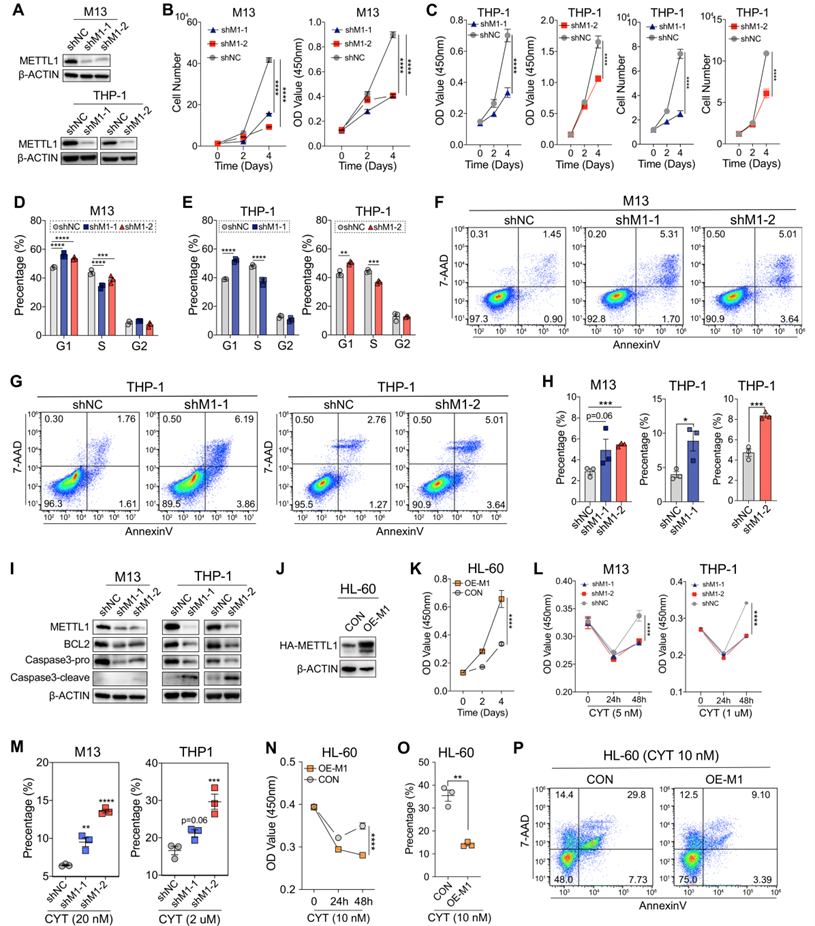
圖二:METTL1敲除抑制人AML細胞的存活和生長。
3.AML中METTL1的表達受DNA甲基化的調控
之前的數據表明,METTL1的表達可能受到DNA甲基化的調控。為了研究METTL1在AML細胞中保持高表達狀態的原因,從數據庫DiseaseMeth和TCGA中收集AML的DNA甲基化陣列數據(450k)發現AML患者中METTL1的DNA甲基化水平(chr12:58165414–58167914)低于健康對照組(DiseaseMeth數據,圖3A)。根據美國國家生物技術信息中心(NCBI)的基因信息,METTL1位于12號染色體的第一外顯子58162254-58165888(-)位點上,該DNA甲基化區域大約位于距離轉錄起始位點(TSS)2千堿基(kb)以內,因此可以歸類為基因啟動子區域。此外,METTL1的DNA甲基化水平與METTL1 mRNA表達呈負相關(TCGA數據,圖3B)。通過應用靶向整體DNA去甲基化水平的基于胞嘧啶的TET酶抑制劑(bobcat339鹽酸)處理AML細胞,發現使用bobcat339鹽酸處理24h和48h后,AML細胞系中METTL1的轉錄水平顯著降低,同時METTL1蛋白下調(圖3C、D),表明Tet家族調控的METTL1基因啟動子區域的DNA去甲基化可能在調控AML細胞中METTL1激活中發揮主要作用。
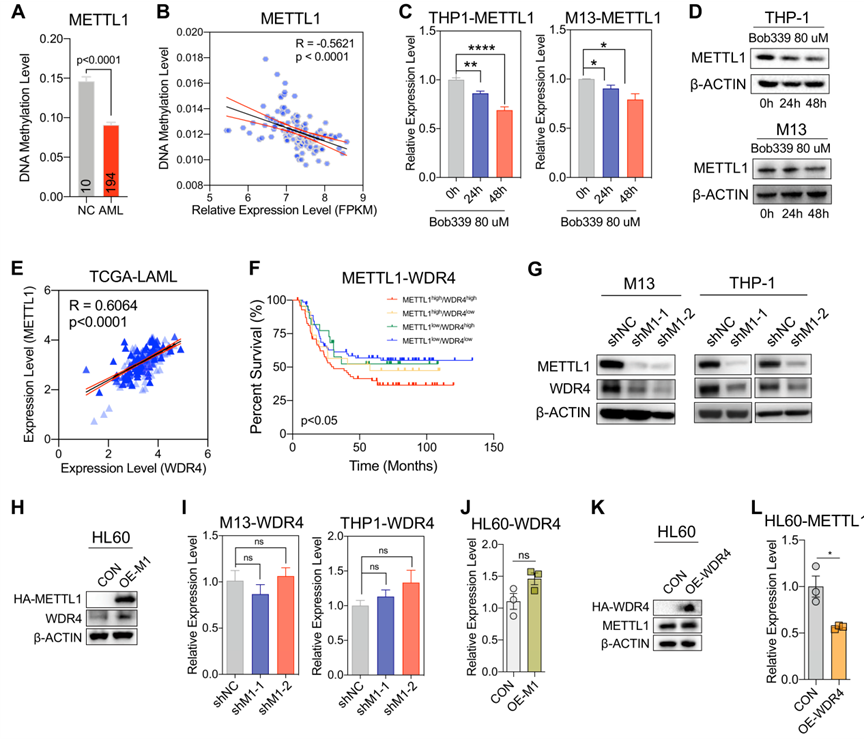
圖三:DNA甲基化調節AML細胞中METTL1的表達以及METTL1和WDR4的相關性。
4.METTL1和WDR4在AML細胞中的協同表達由METTL1引導
之前的研究表明,METTL1和WDR4共同作用以增加RNA中的m7G修飾,并在包含AML在內的各種癌癥中顯示出協同表達模式。為了進一步研究AML中METTL1和WDR4協同表達的調控機制,首先分析了癌癥基因組圖譜(TCGA)中的急性髓系白血病(LAML)轉錄組數據集,并確認METTL1的轉錄水平與WDR4呈正相關(r=0.6064,p<0.0001,圖3E)。此外,同時表達高METTL1和WDR4的AML患者預后最差(圖3F),表明METTL1和WDR4之間存在正協同表達模式。進一步分析表明,WDR4的蛋白水平在METTL1敲除后顯著降低,并隨著METTL1過表達而增加(圖3G,H)。然而,WDR4的mRNA表達水平在AML細胞中METTL1敲除或過表達后沒有明顯變化(圖3I,J),表明WDR4通過METTL1的調控可能發生在轉錄后水平。值得注意的是,在AML細胞中,WDR4表達的改變對METTL1的表達水平無明顯影響,無論是在mRNA水平還是在蛋白水平(圖3K,L)。這些結果表明,METTL1和WDR4在AML細胞中的協同表達是由METTL1通過轉錄后調控機制引導的。
5.敲除METTL1會導致細胞中tRNA和mRNA上的m7G修飾豐度降低
為了證實METTL1是否能決定AML細胞中tRNA和mRNA上m7G修飾的豐度,應用基于LC-MS/MS的高通量RNA修飾定量檢測平臺檢測METTL1敲除AML細胞中m7G RNA修飾的狀態。首先,從有METTL1或沒有METTL1的AML細胞中分離出富含tRNA的片段。通過LC-MS/MS平臺,對tRNA和mRNA上的18種RNA修飾進行量化(圖4A).與對照組相比,METTL1基因敲除的AML細胞中tRNA上的m7G水平明顯下降,而其他類型的RNA修飾的豐度幾乎不受影響(圖4B-D),表明METTL1會特異性地影響AML細胞中tRNA上的m7G豐度。最近的研究表明,哺乳動物細胞中的mRNA也含有相當豐富的內部m7G修飾,因此我們接下來要檢測富含poly(A)的mRNA上的m7G水平。由于mRNA僅占總RNA的一小部分(約3–7%),而mRNA通常在每個分子的5'末端都含有三個m7G帽,因而進行了兩輪poly(A)富集實驗以去除其他RNA的污染,并進行了兩次去帽實驗以在很大程度上去除mRNA 5'末端的m7G帽,從而聚焦于mRNA的豐度變化。結果表明,在METTL1敲除的AML細胞中,poly(A)富集的mRNA上的m7G水平明顯下降,,而m2G,m1G,m22G,m227G不受影響(圖4E-G)。有趣的是,與tRNA上的RNA修飾豐度不同,在METTL1敲除后,在METTL1敲除的AML細胞中,poly(A)富集的mRNA上的m7G水平明顯下降,表明在METTL1敲除的AML細胞中,poly(A)富集的mRNA上的m7G水平明顯下降。
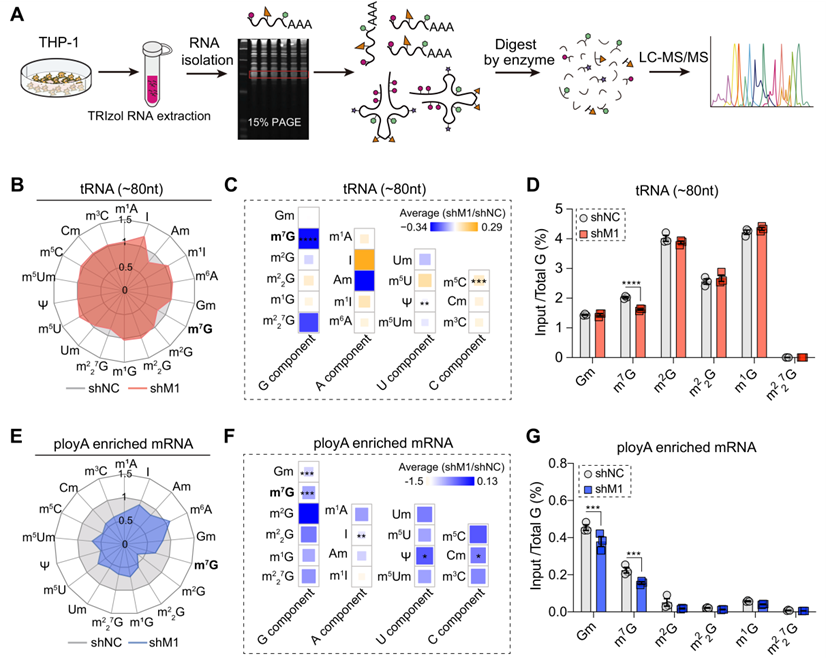
圖四:敲除METTL1會導致THP-1細胞中tRNA和mRNA上的細胞m7G修飾豐度降低
6.METTL1敲除通過tRNA表觀轉錄組失調減少AML細胞中新生蛋白質合成
最近的研究表明,缺失METTL1可降低哺乳動物細胞中m7G修飾轉錄本的翻譯效率,并強調mRNA內部m7G是一種新型表轉錄組標記,在翻譯中具有調控作用。此外,研究還表明,METTL1/WDR4介導的tRNA甲基組上的m7G也是mESC中正常mRNA翻譯所必需的,并調控ESC的自我更新和分化。由于數據顯示METTL1的敲除可降低AML細胞中富含poly(A)的mRNA和總tRNA上的m7G水平,接下來要研究的是METTL1敲除引起的AML細胞中m7G豐度的降低是否會影響細胞翻譯效率。由此進行了OPP標記實驗,以檢測AML細胞中新生蛋白質的整體生物發生情況。結果顯示,通過siRNA敲除METTL1確實降低了AML細胞中OPP平均熒光強度(MFI)的水平(圖5A,B),表明在METTL1敲除的AML細胞中,新生蛋白合成和全局mRNA翻譯受到了抑制。然而,轉錄組測序顯示METTL1敲除的AML細胞中幾乎沒有基因發生改變(圖5C),這一結果表明,在METTL1敲除的AML細胞中觀察到的新生蛋白合成抑制可能并不是主要受METTL1敲除的AML細胞中的基因改變影響(圖5D)。此外,定量蛋白質組學實驗發現,METTL1敲除誘導下調的蛋白富集于蛋白翻譯過程和一系列酶活性(圖5D、E),這與上述結果一致,表明METTL1調控AML細胞的全局翻譯。
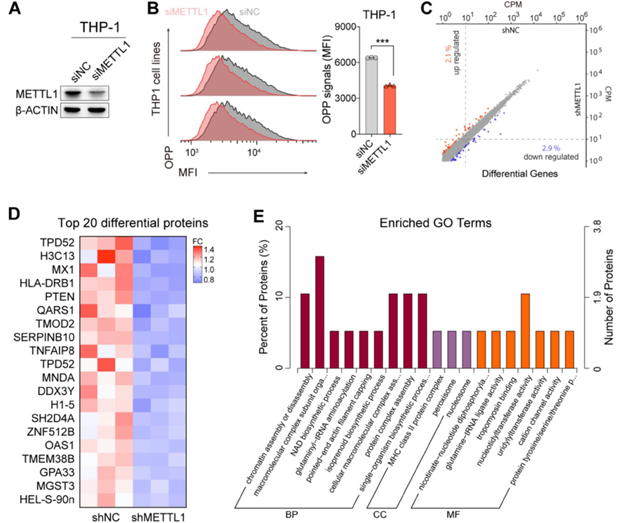
圖五:敲除METTL1可抑制THP-1細胞中新生蛋白質的合成
為了進一步證實METTL1敲除AML細胞的翻譯抑制是否是由m7G失調的tRNA甲基組誘導的,首先對AML細胞中一些選定的tRNA進行Northern印跡檢測,數據顯示與METTL1對照AML細胞相比,METTL1敲除AML細胞中tRNAAla、tRNAVal和tRNALeu的水平顯著下降(圖6A-D)。表明METTL1介導的tRNA m7G缺失可能會影響AML細胞中的細胞tRNA譜。進一步的OPP標記實驗顯示,與METTL1對照細胞相比,轉染了從METTL1敲除細胞中分離的tRNA的AML細胞的OPP平均熒光強度(MFI)水平降低(圖6E-G),表明m7G失調的tRNA甲基化組也影響了細胞tRNA庫的翻譯功能。綜上所述,上述結果表明,METTL1介導的tRNA m7G修飾可調控細胞tRNA庫和tRNA表轉錄組,從而進一步導致AML細胞全局mRNA翻譯失調。
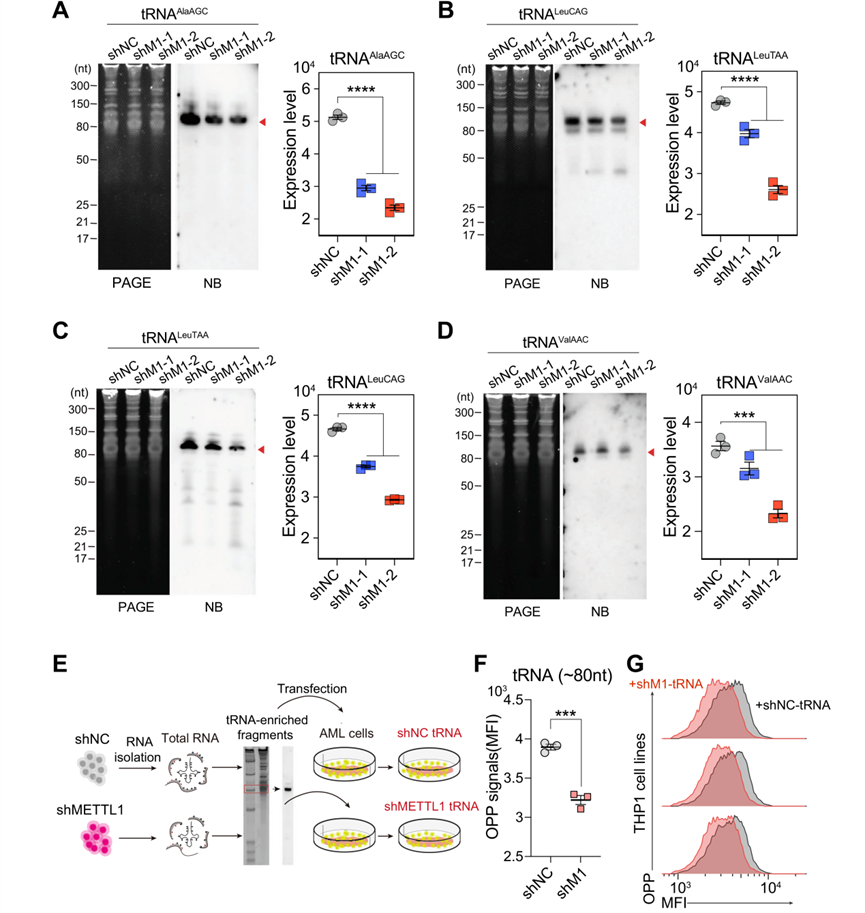 圖
圖
六:敲除METTL1會導致tRNA失調,從而抑制新生蛋白質的合成
7.METTL1促進AML細胞中tRNA衍生小RNA的生物發生
為了研究METTL1基因的敲除如何影響tRNA譜,用RNase A/T1處理從METTL1敲除的AML細胞和METTL1對照AML細胞中分離的總tRNA。結果表明,METTL1的敲除增加了tRNA對Rnase A/T1降解的敏感性(圖7A),從而解釋了在AML細胞中觀察到的由METTL1敲除引起的tRNA改變。作為一種新型的調節性小非編碼RNA,tRNA衍生的小RNA—tsRNA在各種基本生物學條件下以及調節多種癌癥的腫瘤發生中發揮重要作用。tsRNA的生物發生對各種細胞應激敏感,由其前體tRNA的二級結構和甲基化組決定,可將切割位點暴露于多個Rnase,促進tsRNA的生物發生。在這項研究中發現METTL1的敲除降低了總tRNA中的m7G水平,促進了Rnase A/T1消化下的tRNA降解,這可能會促進tsRNA在AML細胞中的生物發生。為了探究敲除METTL1是否能提高AML細胞中tsRNA的整體水平進行了METTL1敲除和METTL1對照AML細胞之間的高通量小非編碼RNA測序實驗,結果表明,在METTL1敲除AML細胞中,包括5'tsRNA、內在tsRNA和3'CCA-tsRNA在內的一些tsRNA的水平顯著升高(圖7B),通過Northern印跡和RT-PCR進一步證實了這一點(圖7C-E)。相反,高通量小非編碼RNA測序和RT-PCR實驗表明,METTL1過表達的AML細胞中大多數tsRNA相應減少(圖7F、G)。然而,當將來自METTL1敲除細胞的分離的tsRNA片段轉染到AML細胞中時,AML細胞中的整體翻譯效率并未受到影響,這表明tsRNA的分子調控機制可能不像AML細胞中的tRNA那樣參與細胞新生蛋白質合成,應進行更多研究以揭示tsRNA在AML白血病發生中的作用機制。總之,上述結果表明,METTL1介導的tRNA上的m7G修飾在調控AML細胞中tRNA穩定性和tsRNA生物發生中起著重要作用,可能會進一步影響AML的細胞活力和白血病發生。
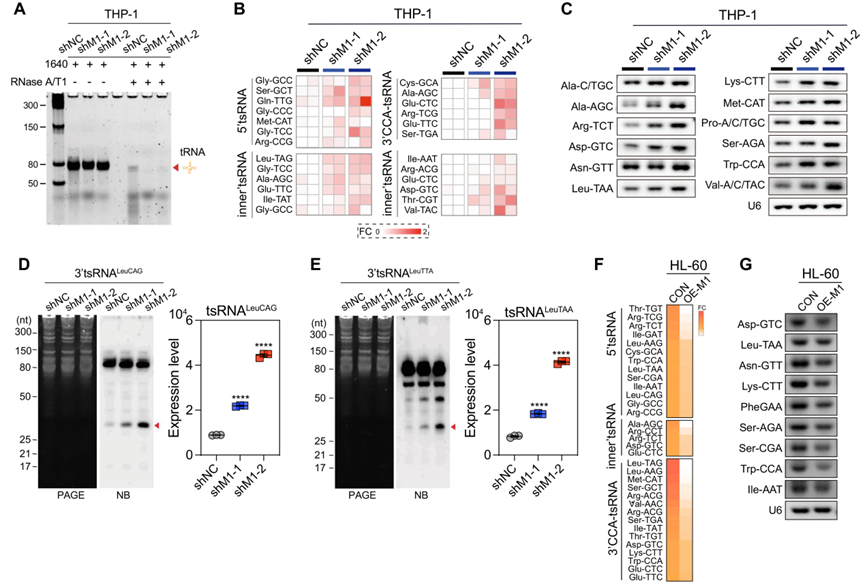
圖七:METTL1敲除促進tsRNA生物發生。
實驗方法:
qRT-PCR,WB,生物信息學分析,CCK-8法和細胞計數法,northern印跡法、液相色譜-串聯質譜(LC-MS /MS)、tRNA穩定性分析、轉錄組測序、小非編碼RNA測序、定量蛋白質組學和蛋白質合成測定。
參考文獻:
Zhao P, Xia L, Chen D, Xu W, Guo H, Xu Y, Yan B, Wu X, Li Y, Zhang Y, Zhang X. METTL1 mediated tRNA m7G modification promotes leukaemogenesis of AML via tRNA regulated translational control. Exp Hematol Oncol. 2024 Jan 24;13(1):8. doi: 10.1186/s40164-024-00477-8. PMID: 38268051; PMCID: PMC10807064.