






ZDHHC5對下丘腦小膠質細胞PKCδ的棕櫚酰化作用是脂肪肝治療靶點
下丘腦通過神經內分泌信號在控制脂質代謝方面發揮重要作用。然而,目前在下丘腦中還沒有能夠有效改善人類脂質代謝的藥物靶點。在這項研究中,作者發現抗瘧藥物蒿甲醚(ART)通過特異性抑制高脂飲食誘導小鼠下丘腦中的小膠質細胞活化,顯著改善脂質代謝。從機制上講,ART保護小膠質細胞周圍的促甲狀腺激素釋放激素(TRH)神經元免受炎癥損傷,并促進TRH釋放到外周循環中。因此,TRH刺激甲狀腺激素(TH)的合成,從而顯著改善肝臟脂質紊亂。隨后,作者使用生物素標記的ART化學探針來鑒定小膠質細胞中的直接細胞靶點為蛋白激酶Cδ(PKCδ)。重要的是,ART直接靶向PKCδ,通過阻斷鋅指DHHC型棕櫚酰轉移酶5(ZDHHC5)的結合來抑制其棕櫚酰化修飾,從而抑制下游神經炎癥信號傳導。在體內,下丘腦小膠質細胞特異性PKCδ敲低顯著損害了小鼠ART依賴性神經內分泌調節和脂質代謝改善。此外,人腦組織中的單細胞轉錄組學分析顯示,小膠質細胞中PKCδ的水平與高脂血癥患者呈正相關,從而突出了臨床轉化價值。總之,這些數據表明,下丘腦中小膠質細胞PKCδ的棕櫚酰化通過下丘腦-肝臟通訊調節外周脂質代謝,并為脂肪肝疾病提供了一個有前景的治療靶點。該研究于2024年1月發表在《Theranostics》,IF 12.4。
技術路線:

主要研究結果:
1 藥物再定位確定抗瘧藥物ART是一種降脂劑
藥物重新定位是開發代謝性疾病新療法的一種很有前途的策略,從而改善傳統藥物的開發。HFD喂養的小鼠被用作研究脂質代謝的實驗模型,特別關注脂質代謝紊亂。為了探討抗瘧藥物ART是否能減輕HFD誘導的脂質代謝紊亂,C57BL/6J小鼠接受對照飲食或HFD治療5周(圖1A)。如圖1B-1D所示,HFD治療顯著提高了血清總膽固醇(TC)、總甘油酯(TG)和非高密度脂蛋白膽固醇(non-HDL-C)水平,而ART以劑量依賴性方式抑制了這些水平。此外,油紅O染色顯示,HFD處理促進肝中脂滴的積聚,ART顯著逆轉這一現象(圖1E)。H&E染色還顯示ART改善了脂質代謝紊亂相關的脂肪變性(圖1F)。此外,定量實時聚合酶鏈式反應(RT-PCR)分析顯示,在接受ART治療的受試者的肝臟中,脂質代謝相關基因,即肉堿棕櫚酰轉移酶1A(Cpt1a)和過氧化物酶體增殖物激活受體α(Ppara)的表達顯著增加。此外,脂質合成基因的表達,硬脂酰輔酶A去飽和酶1(Scd1),通過HFD治療上調的在口服ART后被觀察到下調(圖1G)。在脂質代謝相關蛋白的情況下觀察到類似的模式。ART的給藥顯著增加了CPT1A和PPARA蛋白的表達,同時抑制了SCD1蛋白的表達。同時,我們發現ART明顯降低了血清谷丙轉氨酶(ALT)和谷氨酰胺草乙酸轉氨酶(AST)水平,并使肝臟超氧化物歧化酶(SOD)、谷胱甘肽過氧化物酶(GSH-PX)活性和丙二醛(MDA)水平正常化,證實明顯的保肝作用(圖1H-1I)。為了驗證ART對脂質代謝紊亂的作用,作者使用另一種Balb/c小鼠來測試之前的觀察結果。在這里,作者發現ART對脂質代謝表現出類似的改善作用,包括血脂水平、肝臟脂質積聚、脂質代謝相關基因表達、蛋白質表達和氧化應激。總之,這些結果表明ART是一種有效的降脂劑,可以改善肝臟脂質代謝紊亂。
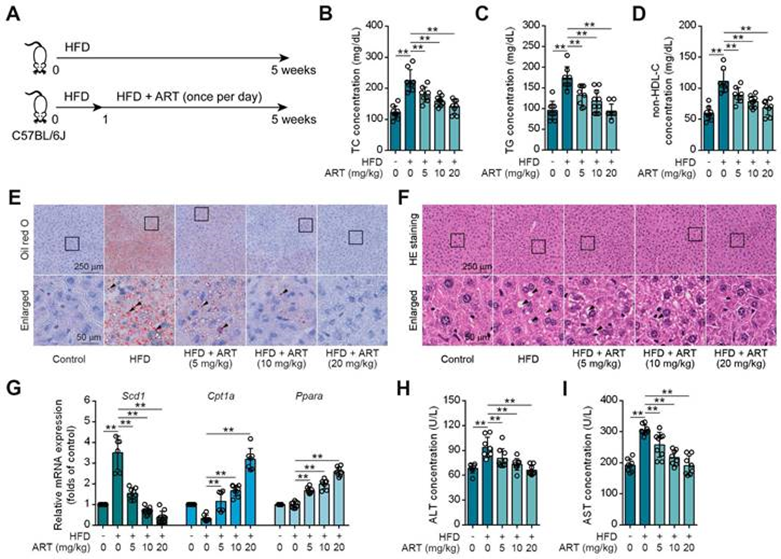
圖1:ART抑制HFD喂養的C57BL/6J小鼠肝臟脂質代謝紊亂
2 ART通過激活神經內分泌系統改善肝臟脂質紊亂
為研究ART如何調節脂質代謝,作者建立了棕櫚酸(PA)或油酸(OA)誘導的人肝細胞(LO2)模型。尼羅紅染色顯示,PA或OA處理明顯促進LO2細胞中的脂質積累,但ART并不能阻止這些變化。類似的觀察結果表明,ART沒有阻斷PA或OA誘導的人HepG2細胞中的脂質積聚。此外,ART對PA誘導的LO2細胞中Cpt1a和Ppara基因表達沒有顯著影響。為進一步深入了解ART對肝臟脂質代謝的影響,作者使用原代肝細胞進行實驗。研究結果顯示,當接受PA或OA治療時,ART不會增強Cpt1a和Ppara基因或蛋白質的表達。因此,這些結果表明ART劑量不能直接調節肝臟脂質代謝。
先前的研究表明,小腸上皮細胞的攝取和吸收與脂質代謝紊亂的發展有關。為闡明ART是否影響小腸上皮細胞的攝取,作者進行了Caco-2轉運實驗。他們發現硬脂酸(SA)、PA、OA和膽固醇(CHO)從心尖(AP)轉運到基底外側(BL),不受ART的影響。這些結果表明ART劑量不干擾腸道吸收。此外,ART對體重和食物攝入量沒有明顯影響。
接下來,作者尋找ART介導的降脂作用的其他潛在機制。據報道,神經內分泌系統在調節能量代謝中起著至關重要的作用,因此作者假設ART通過調節激素分泌改善脂質代謝。首先,作者檢測了脂質代謝相關激素的釋放,包括腎上腺素、胰島素和胰高血糖素。在這里,ART并沒有顯著調節這些激素的水平。此外,研究結果顯示,在接受ART治療后,喂食HFD小鼠PVN區域的TRH水平顯著增加(圖2A)。此外,作者的研究還表明,ART治療導致血清TSH、游離總甲狀腺素(FT4)和游離總三碘甲狀腺原氨酸(FT3)水平顯著上調。這些觀察結果表明,ART可以通過調節神經內分泌系統來改善HFD誘導的脂質代謝紊亂(圖2B-2D)。特別地,在LO2細胞上進行了使用l-THY作為合成甲狀腺激素的概念驗證實驗。發現l-THY顯著抑制PA或OA誘導的脂質積聚。類似地,l-THY減少了PA或OA誘導的HepG2細胞中的異常脂質積聚,表明TH在控制肝臟脂質代謝中的重要作用。
為進一步證實ART介導的TH釋放和脂質代謝的相關性,作者接下來使用噻唑(THI,一種甲狀腺功能抑制劑)治療HFD喂養的小鼠(圖2E)。如圖2F-2H所示,THI顯著逆轉了ART調節的TC、TG和非HDL-C的下降。此外,THI阻斷了ART介導的對肝臟脂質積聚的抑制(圖2I-2J)。同時,作者發現THI明顯消融ART降低了Scd1,而ART增加了Cpt1a、Ppara mRNA水平。
由于甲狀腺激素受體(THRs)是能量消耗和肝臟脂質代謝的重要調節因子,作者試圖探討ART誘導的TH釋放是否通過TH/THRs途徑改善肝臟脂質代謝。在這項研究中,開發了一種肝特異性腺相關病毒-5-小分子短病毒蛋白-RNA-TH受體β(Ash-Thrb),可特異性靶向并降低THRβ蛋白的表達。作為Thrb敲低的結果,THRβ蛋白表達被成功降低(圖2K)。數據表明,Thrb敲低逆轉了注射Ash-Thrb的C57BL/6J小鼠中ART-介導的TC、TG和非HDL-C的下調(圖2L-2N)。此外,作者的研究結果表明,在Thrb敲低模型中,ART治療對肝臟脂質積聚沒有表現出顯著的抑制作用(圖2O-2P)。這一觀察結果表明,TH/THRs信號通路在ART調節肝臟脂質紊亂中起著至關重要的作用。此外,作者還利用HepG2細胞研究了TH介導的脂質代謝的潛在信號機制。作者發現,在HepG2細胞中,l-THY通過阻斷核受體輔壓因子1(NcoR1)和組蛋白脫乙酰酶3(HDAC3)之間的相互作用,顯著降低PA誘導的乙酰組蛋白H3(K9)和乙酰組蛋白H4(K16),HDAC3先前被報道為肝臟脂質代謝的基本生物途徑。此外,Thrb敲低抑制了HepG2細胞中l-THY改善的Cpt1a和Ppara mRNA水平,表明TH通過THR依賴性的乙酰組蛋白下調來調節脂質代謝。總之,這些數據表明,ART可能通過TH/THR級聯調節肝臟脂質代謝(圖2Q)。
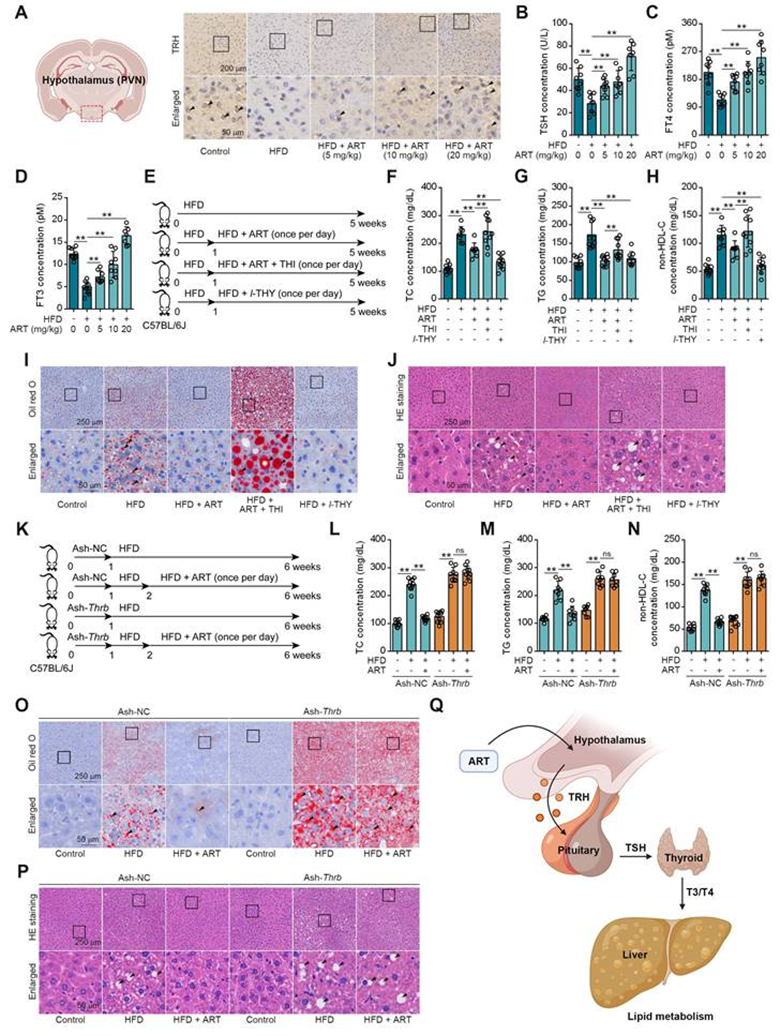
圖2:TH介導ART治療C57BL/6J小鼠HFD喂養的肝臟脂質代謝紊亂
3下丘腦小膠質細胞介導的神經炎癥對TRH神經元損傷的神經內分泌控制作用
下丘腦TRH神經元對甲狀腺激素水平的中樞調節已被廣泛認可。由于小膠質細胞的激活與神經炎癥和由此產生的神經元損傷高度相關,作者推測ART可能通過抑制HFD誘導的小膠質細胞激活來保護下丘腦TRH神經元。正如預期的那樣,作者在HFD喂養的小鼠下丘腦PVN中觀察到高電離鈣結合適配器分子1(Iba1)的表達(小膠質細胞的標志物)(圖3A),表明脂質代謝紊亂加速了小膠質細胞介導的神經炎癥。然而,ART顯著降低Iba1的表達并改善了小膠質細胞的形態(圖3A)。同時還發現,ART降低了HFD誘導的下丘腦PVN中的小膠質細胞炎癥介質(腫瘤壞死因子-α、TNF-α和白細胞介素-6、IL-6)(圖3B-3C)。PVN區域的免疫熒光分析顯示TNF-α/IL-6與Iba1共定位,表明小膠質細胞主要參與這些炎癥因子的產生。此外,作者發現ART顯著阻斷PA誘導的BV-2和原發性小膠質細胞中一氧化氮(NO)、TNF-α和IL-6的產生(圖3D-3F)。
鑒于小膠質細胞在神經炎癥誘導的神經元損傷中發揮著重要作用,作者建立了原代神經元-小膠質細胞共培養的實驗模型(圖3G)。研究結果顯示,來源于PA處理小膠質細胞的條件培養基(CM-PA)顯著降低來源于PVN神經元的活力,并抑制TRH的表達。值得注意的是,ART直接治療并沒有減輕這種有害影響。然而,如圖3H-3I所示,CM-PA治療降低神經元活力和TRH表達,而PA/ART聯合治療的小膠質細胞衍生條件培養基(CM-ART)顯著增加了神經元活力。在體內,作者對PVN中的TRH和Iba1進行免疫熒光染色。觀察到活化的小膠質細胞(Iba1陽性)周圍的TRH陽性神經元數量顯著減少,ART治療有效改善了這一情況(圖3J)。此外,TRH和膠質纖維酸性蛋白(GFAP)的免疫熒光染色顯示,ART對TRH和GFAP(星形膠質細胞的標志物)的共同表達沒有表現出明顯的影響(圖S6Q)。總之,這些數據表明,小膠質細胞介導的神經炎癥在下丘腦PVN中ART保護的TRH神經元中起著至關重要的作用。
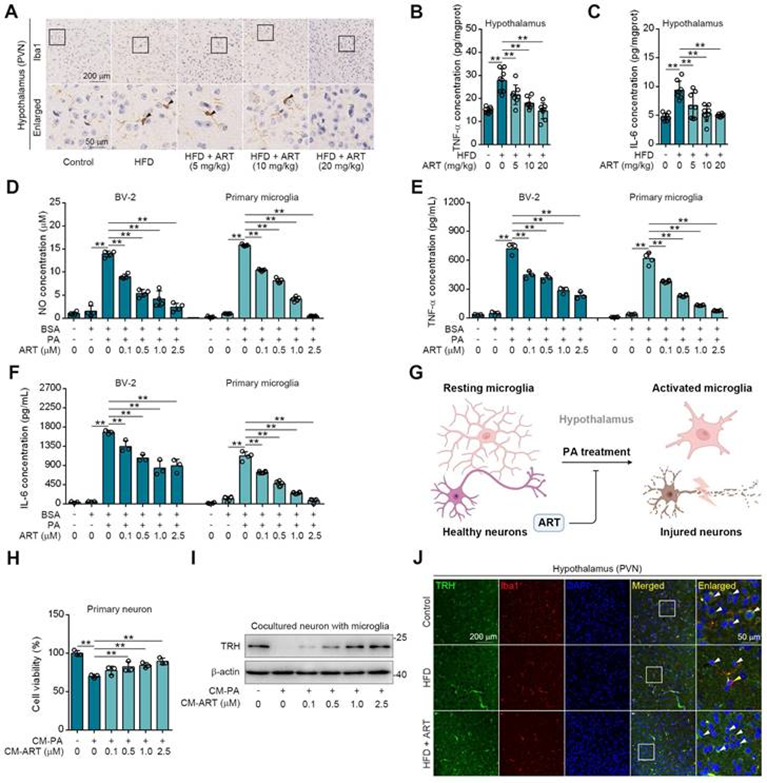
圖3:ART保護PVN區神經元免受神經炎癥的影響
4 PKCδ作為細胞靶點對抗微膠質細胞活化
為探索小膠質細胞介導的神經炎癥靶蛋白,作者進行了熱蛋白質組分析(TPP)技術來鑒定ART的細胞靶點。然后,通過高效液相色譜-串聯質譜(HPLC-MS/MS)總共鑒定了2292個潛在的ART結合蛋白,根據實驗重復性(重/輕,H/L比)和p值確認了其中118個(圖4A)。從最初的候選蛋白庫中,基于它們對轉錄表達譜的顯著貢獻,共選擇20種潛在的靶蛋白進行進一步篩選。這一選擇過程涉及利用轉錄組學與連接性圖分析相結合。接下來,生物功能注釋揭示了三種主要的炎癥相關蛋白,包括蛋白激酶Cδ(PKCδ)、Serpinb1a和Serpinb6。特別是,PKC在先前的報告中強調了一種有前景的“可藥用性”,暗示ART可能是一種潛在的PKC抑制劑。接下來,用泛PKC抑制劑staurosporine(STAU)進行的NO測定也驗證PKC是與神經炎癥病理相關的關鍵靶點(圖4B)。
PKC家族成員分為多種亞型,負責不同的生物功能。除了PKCδ,還報道了PKCα、PKCζ和PKCμ與炎癥過程高度相關。為探索ART與PKCδ的選擇性相互作用,作者合成了生物素標記的ART探針(生物素ART)用于下拉分析。如圖4C-4E所示,生物素ART顯著降低了PKCδ,而生物素ART被過量的ART阻斷。然而,ART與其他PKC沒有表現出顯著的相互作用,包括PKCα、PKCζ和PKCμ。此外,作者還發現ART在細胞熱位移測定(CETSA)實驗中顯著阻止PKCδ蛋白的降解,但對PKCα、PKCζ和PKCμ沒有影響(圖4F)。類似地,藥物親和響應靶標穩定性(DARTS)分析表明,ART專門靶向PKCδ,以抑制其對蛋白酶的蛋白水解(圖4G)。
PKCδ由一個N端調控結構域、一個C端催化結構域和一個短的“hinge”區組成。調控結構域包含C2樣區和C1區,催化結構域包含C3區和C4區。為研究哪一個結構域是ART特別靶向的,作者將PKCδ截短為PKCδ1、PKCδ2、PKCΔ3和PKCδ4(圖4H)。下拉分析顯示,ART與獨特的C1結構域特異性相互作用,但與C2樣、C3或C4結構域無關(圖4I-4J)。總之,作者的數據表明PKCδ是小膠質細胞中ART的重要細胞靶點。
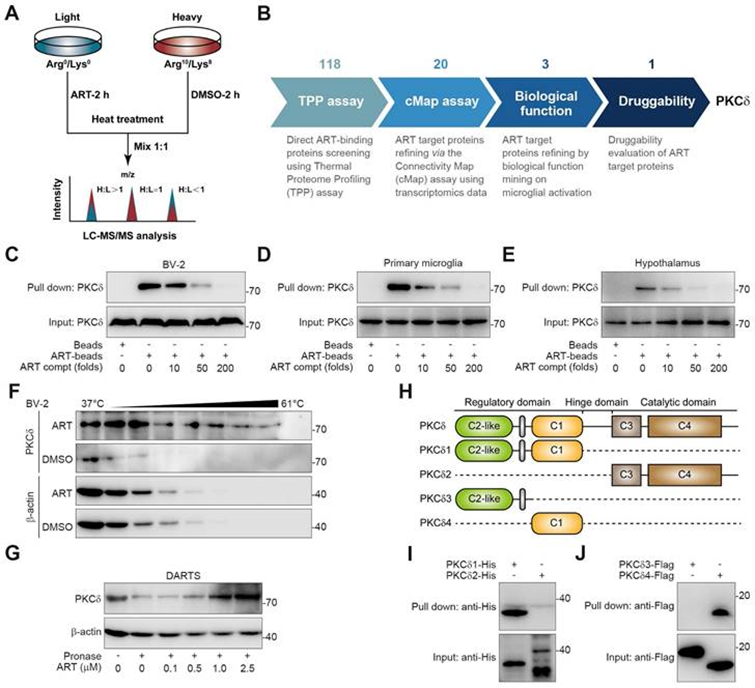
圖4:在BV-2細胞中發現PKCδ作為ART的主要靶點
5 棕櫚酰化修飾PKCδ對小膠質細胞的激活作用
由于PKCδ激活是神經炎癥的關鍵先決條件,作者隨后探討了ART對PKCδ功能的調節機制。最近的研究表明,蛋白質棕櫚酰化在磷酸化調節中起著重要作用,PA與半胱氨酸殘基的共價連接(S-棕櫚酰化)是蛋白質的廣泛修飾。首先,作者觀察到PA治療顯著增加了小膠質細胞中PKCδ的磷酸化,這被ART逆轉(圖5A)。在這里,作者推測PKCδ磷酸化可能通過其先前的棕櫚酰化而受到潛在調節。為此,作者進行酰基生物素交換分析,并觀察到PA顯著增加了小膠質細胞中PKCδ棕櫚酰化,這被ART逆轉(圖5B)。此外,在喂食HFD小鼠的下丘腦中觀察到PKCδ棕櫚酰化明顯增加。特別是,發現下丘腦中的PKCδ棕櫚酰化被ART治療顯著阻斷。接下來,作者使用2BP,一種蛋白質棕櫚酰化阻滯劑,來抑制PA誘導的BV-2細胞中的PKCδ棕櫚酰化。如圖5C所示,2BP幾乎完全消除了ART介導的PKCδ磷酸化抑制。此外,作者觀察到2BP治療顯著逆轉了ART介導的NO、TNF-α和IL-6釋放的抑制作用(圖5D-5F),表明PKCδ棕櫚酰化在脂質誘導的小膠質細胞活化和神經炎癥中起著至關重要的作用。
為研究PKCδ棕櫚酰化的分子機制,作者進行了全局蛋白質組學分析,并通過HPLC-MS/MS鑒定ZDHHC5是下丘腦中最重要的棕櫚酰轉移酶。然后敲低ZDHHC5,發現PA誘導的PKCδ棕櫚酰化在敲低ZDHHC5的細胞中顯著減弱。同時,ART介導的對PKCδ磷酸化的抑制也被Zdhhc5敲低顯著阻斷(圖5G),表明Zdhhc5依賴的PKCδ棕櫚酰化有助于PKCδ磷酸化。Co-IP實驗還顯示,PKCδ-ZDHHC5相互作用可被ART在藥理學上抑制(圖5H)。此外,在si-Zdhhc5治療后,ART失去了對NO、TNF-α和IL-6釋放的抑制作用(圖5I-5K)。因此,這些結果表明,ZDHHC5依賴性棕櫚酰化在小膠質細胞中高度參與PKCδ的激活(圖5L)。
接下來作者研究PKCδ依賴性神經炎癥信號通路。KEGG通路分析富集了3種主要的炎癥信號通路,包括NLRP3、NF-κB和Janus激酶信號轉導轉錄激活劑(Jak-Stat)。接下來,進行蛋白質印跡以研究ART對NLRP3炎癥小體途徑的影響。PA增加了NLRP3、胱天蛋白酶1、IL-1β和IL-18的水平,這一水平被ART顯著逆轉。此外,PA組κB激酶β磷酸化抑制劑(pIKKβ)、pNF-κB p65和核因子κBα磷酸化抑制物(pIκBα)的表達顯著增加,這被ART抑制。此外,作者觀察到PA誘導Jak2、Stat5和Stat3磷酸化水平的增加,這被ART治療阻斷。總之,這些數據表明,PKCδ通過獨特的棕櫚酰化修飾作為小膠質細胞活化和神經炎癥的脂質傳感器。
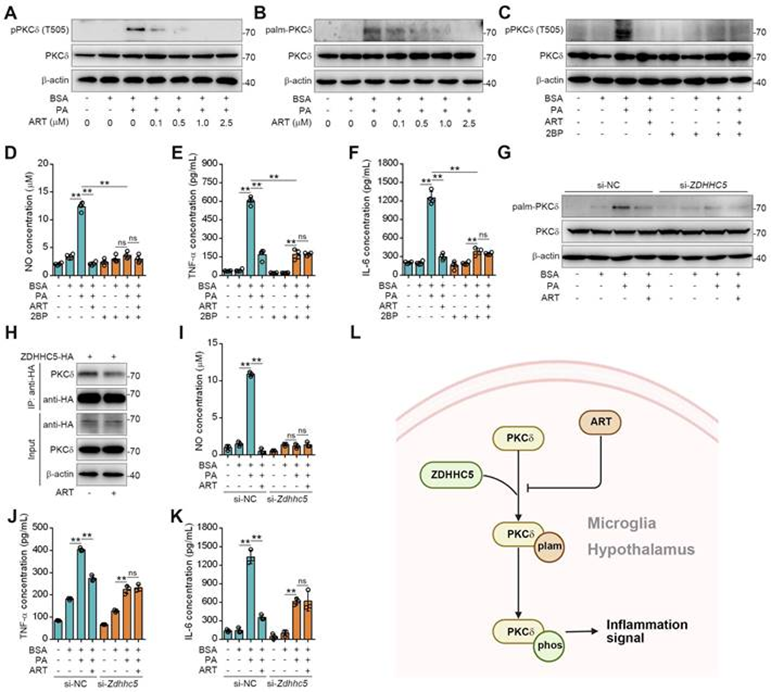
圖5:ART通過阻斷PKCδ棕櫚酰化抑制PKCδ磷酸化以減輕神經炎癥
6藥理學靶向下丘腦微膠質細胞PKCδ減輕體內肝臟脂質代謝紊亂
首先,作者研究了藥物靶向下丘腦PKCδ是否表現出抗神經炎癥作用。通過CRISPR/Cas9建立Prkcd+/-小鼠模型。發現ART對HFD喂養的Prkcd+/+小鼠PVN中Iba1的表達產生了明顯的抑制作用,這在Prkcd+/-小鼠中顯著逆轉。此外,ART阻斷了HFD喂養的Prkcd+/+小鼠PVN中TNF-α和IL-6的釋放,但在Prkcd+/-小鼠中表現出較小的作用。此外,對ART的血腦屏障(BBB)轉運的藥代動力學分析顯示,ART可以通過血腦屏障并在下丘腦中積累(S10H)。這些數據表明,ART可能通過靶向PKCδ在下丘腦發揮抗神經炎癥作用。
為進一步證實ART是否通過選擇性靶向PVN小膠質細胞中的PKCδ來抑制下丘腦神經炎癥,設計了一種小膠質細胞特異性腺相關病毒-5-小分子短發夾RNA-Prkcd(Ash-Prkcd),用于小鼠PVN的立體定向顱內注射(圖6A-6B)。Ash-Prkcd注射液的給藥有效降低了位于PVN區域小膠質細胞中PKCδ的表達。同樣,作者還發現,在Ash陰性對照(Ash-NC)注射組中,ART有效降低了HFD誘導的Iba1產生,而在Ash-Prkcd注射組中ART對其沒有明顯影響(圖6C)。此外,ART顯著降低Ash NC注射組的下丘腦炎癥介質,包括TNF-α和IL-6,這在Ash Prkcd注射小鼠中得到了有效逆轉(圖6D-6E)。總之,這些數據表明,ART在體內選擇性靶向下丘腦中的小膠質細胞PKCδ,以抑制神經炎癥。
然后,作者試圖測試藥物靶向PKCδ是否具有降脂作用。如圖6F-6I所示,ART顯著增加NC注射PVN中TRH神經元數量,從而促進甲狀腺激素的產生。然而,在注射Ash-Prkcd的小鼠中沒有觀察到類似的效果。此外,在注射Ash-NC的小鼠中,ART顯著改善了HFD誘導的TC、TG和非HDL-C水平,而在注射Ash-Prkcd的小鼠中ART治療沒有顯示出類似的治療效果(圖6J-6L)。同時,ART改善了注射Ash-NC的HFD誘導的小鼠肝臟脂肪變性和脂質代謝相關基因的表達;然而,Ash-Prkcd注射逆轉了ART依賴性治療效果(圖6M-6O)。此外,ART降低了注射Ash NC小鼠的ALT和AST水平,這在注射Ash Prkcd小鼠中也被消融(圖6P-6Q)。為了排除PKCδ對肝細胞脂質代謝的直接影響,作者下調了HepG2和LO2細胞中PKCδ的表達,發現ART對脂質積累沒有明顯的調節作用。總之,作者的研究結果表明,下丘腦中小膠質細胞PKCδ是緩解體內肝臟脂質紊亂的關鍵靶點。
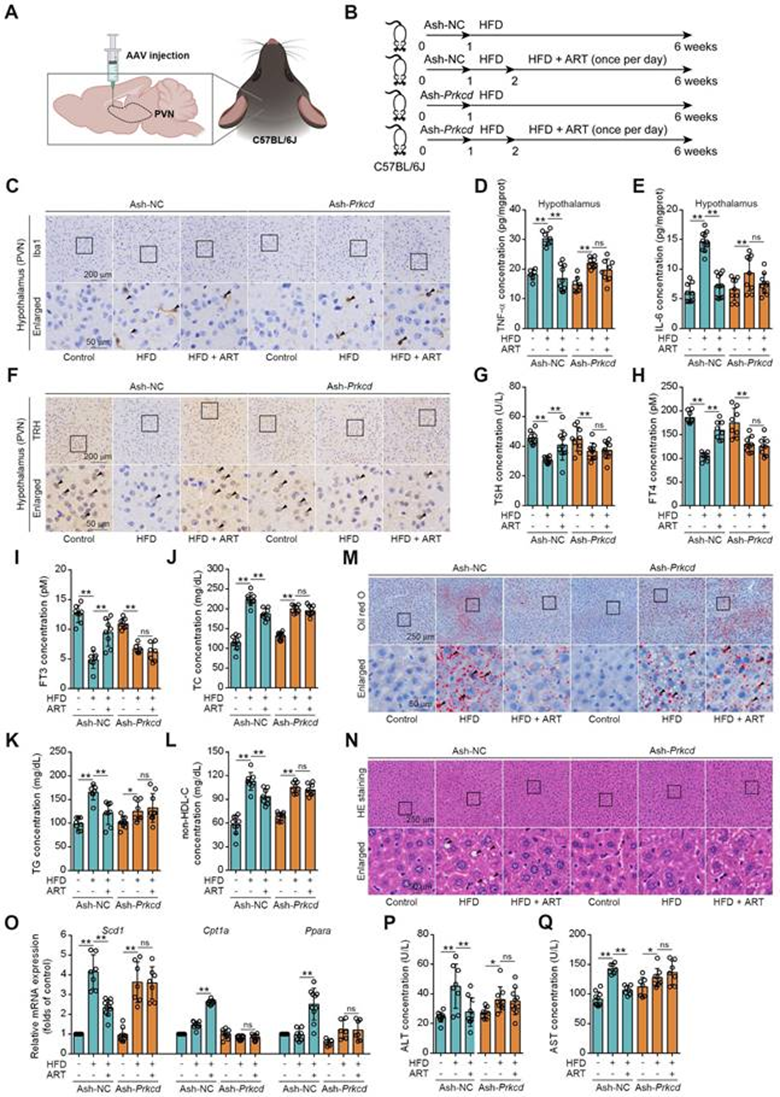
圖6:Prkcd敲低PVN區逆轉ART對C57BL/6J小鼠HFD喂養的肝臟脂質代謝紊亂的抑制作用
7 PKCδ與高脂血癥患者腦內微膠質細胞活化有關
作者從5名對照捐贈者和5名高脂血癥患者那里獲得了腦組織和血清樣本(圖7A)。免疫組織化學和蛋白質印跡結果顯示,高脂血癥患者大腦中的PKCδ、pPKCδ和plam PKCδ水平均較高(圖7B-7C)。此外,免疫熒光分析顯示,在高脂血癥的大腦中,pPKCδ與Iba1的共定位顯著增加。這一發現表明PKCδ激活和小膠質細胞激活之間存在正相關(圖3D)。同時,作者證實了高脂血癥大腦中TRH水平的降低(圖7E)。重要的是,作者還觀察到一種趨勢,但沒有統計學意義,因為高脂血癥患者的TC、TG和LDL-C水平與Iba1和plam PKCδ表達呈正相關的個體間變異性很高,而且受試者數量有限(圖7F)。此外,TRH水平與高脂血癥的嚴重程度呈負相關(圖7F)。總之,這些數據表明PKCδ介導的神經炎癥可能與脂質代謝紊亂有關。
接下來,作者進行了單核RNA-seq,以表征腦組織中的細胞類型。在對照組和患者之間,小膠質細胞和神經元細胞簇的UMAP圖和細胞百分比發生了顯著變化。這些數據顯示,高脂血癥腦小膠質細胞中PKCδ相關炎癥信號持續增加,如Toll樣受體、NF-κB、絲裂原活化激酶樣蛋白(MAPK)、NOD樣受體和JAK-STAT通路(圖7G)。在神經元中,KEGG通路分析表明,幾種常見的神經元存活通路被下調,這些通路與谷氨酸能、膽堿能、GABA能和多巴胺能突觸功能高度相關。此外,一些神經元凋亡途徑,如活性氧信號級聯也被上調。
為進一步對TRH神經元進行詳細的轉錄組學分析,作者在神經元群體中進行了無偏聚類。結果顯示了8個神經元簇(圖7H)。在這些簇中,神經元SAI高表達TRH基因,高脂血癥腦中的細胞百分比顯著降低(圖7I-7J)。對神經元SAI的進一步KEGG分析顯示,TH合成減少,TH信號通路下調,這表明PKCδ介導的小膠質細胞激活可能特別導致高脂血癥腦中TRH神經元損傷(圖7K)。總之,這些數據表明大腦中小膠質細胞PKCδ激活與外周脂質代謝紊亂的臨床關聯。
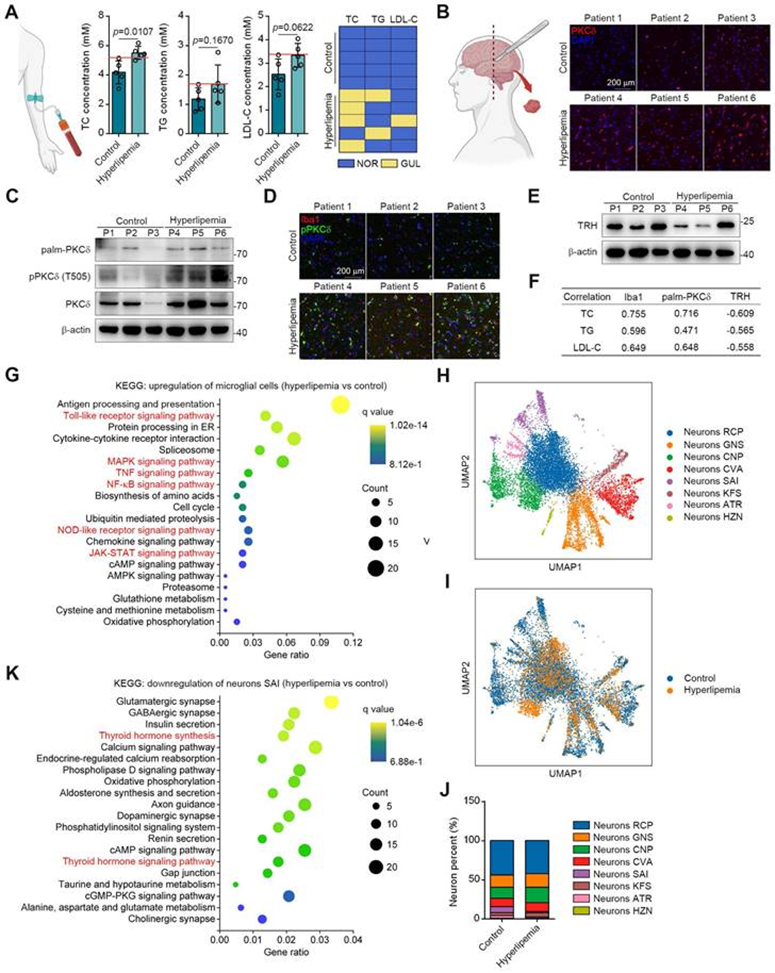
圖7:PKCδ表達在脂肪腦中上調并與高脂血癥進展相關
結論:
總之,作者提供了概念證據,證明下丘腦中的小膠質細胞PKCδ通過神經內分泌信號調節脂質代謝紊亂。特別是,這些發現表明,靶向小膠質細胞PKCδ可能是治療脂肪肝的一種有前途的治療方法,這與目前的治療策略不同。
實驗方法:
細胞培養,油紅O染色,血清生化分析,免疫組化,RT-PCR,尼羅紅染色,免疫熒光,細胞活力分析,熱蛋白質組分析(TPP),單細胞測序,Pull-down,細胞熱位移測定(CETSA),Co-IP,siRNA或質粒轉染,WB,UPLC-MS/MS
參考文獻:
Wang YH, Chen X, Bai YZ, Gao P, Yang Z, Guo Q, Lu YY, Zheng J, Liu D, Yang J, Tu PF, Zeng KW. Palmitoylation of PKCδ by ZDHHC5 in hypothalamic microglia presents as a therapeutic target for fatty liver disease. Theranostics 2024; 14(3):988-1009. doi:10.7150/thno.89602. https://www.thno.org/v14p0988.htm