






EGFR!ALKBH5!防止膠質母細胞瘤中的鐵死亡!
生長因子受體是最重要的致癌途徑之一,但藥物抑制劑作為單一療法的效果有限。在這里,作者發現表皮生長因子受體(EGFR)信號傳導抑制膠質母細胞瘤干細胞(GSCs)中的N6-甲基腺苷(m6A)水平,而遺傳或藥理學EGFR靶向升高m6A水平。激活的EGFR誘導非受體酪氨酸激酶SRC磷酸化m6A去甲基化酶,AlkB同源物5(ALKBH5),從而抑制染色體維持1(CRM1)介導的ALKBH5核輸出,從而允許細胞核中持續的mRNA m6A去甲基化。ALKBH5通過m6A調節和YTH N6 -甲基腺苷RNA結合蛋白(YTHDF2)介導的谷氨酸-半胱氨酸連接酶修飾子亞基(GCLM)的衰變,精密調節鐵死亡。ALKBH5的藥理學靶向增強了EGFR和GCLM抑制因子的抗腫瘤功效,支持EGFR-ALKBH5-GCLM的致癌軸。總的來說,EGFR通過ALKBH5去甲基化酶的核保留對表轉錄組景觀進行重編程,以防止鐵死亡,為致命癌癥的治療提供了治療范例。本文于2023年11月發表于《Molecular cell》, IF: 16.0,Q1。
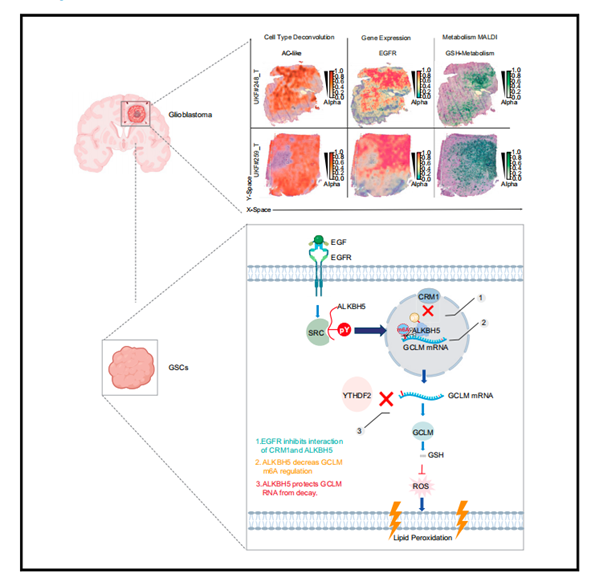 作用機制圖解
作用機制圖解
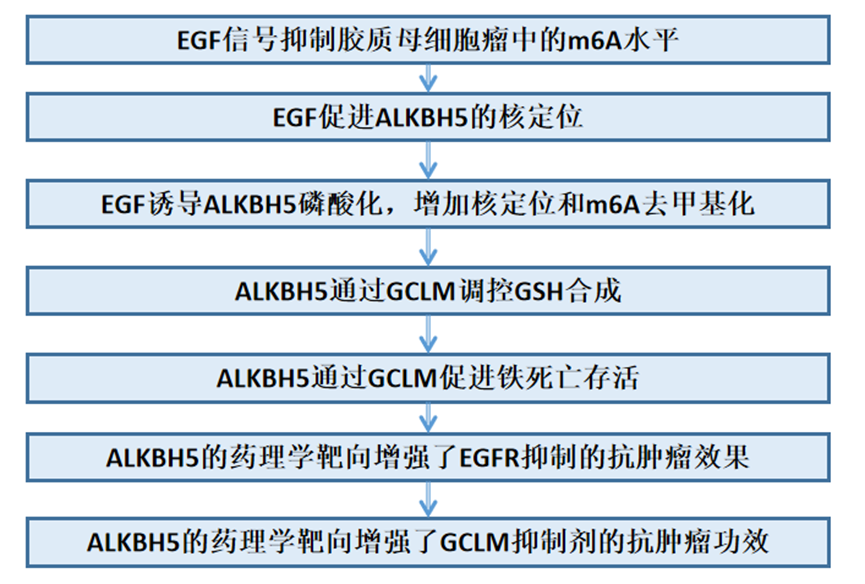
技術路線:
主要實驗結果:
1、EGF信號抑制膠質母細胞瘤中的m6A水平
作者最近報道了m6A寫入者和擦除者產生的基因表達特征與GSCs中選擇的RTK通路(特別是PDGFR、VEGFR和EGFR)相關。PDGFR通過轉錄誘導METTL3增加m6A水平,而VEGF不改變m6A水平,提示RTKs在表轉錄組學中具有不同的機制。EGFR和PDGFR是膠質母細胞瘤中兩個重要的致癌基因,它們與不同的轉錄亞型有關,但兩者在腫瘤內經常發生改變,顯示出腫瘤內的空間異質性。利用最近報道的膠質母細胞瘤的空間多組學分析提供了腫瘤組織學和細胞類型分布(圖1A)。膠質母細胞瘤中GFR的空間表達分布顯示EGFR和PDGFRA是互斥的(圖1B)。EGFR與徑向膠質細胞壁龕和星形細胞樣(AC樣)亞型相關,而PDGFR與少突細胞祖細胞樣(OPC樣)壁龕和神經祖細胞樣(NPC樣)亞型相關(圖1C、1D)。基于這一背景,作者假設EGFR通過與PDGFR不同的機制調控m6A水平。EGFR在膠質母細胞瘤中穩定m6A解讀子YTHDF2,但在肝細胞癌中,YTHDF2通過破壞EGFR mRNA的穩定性來降低腫瘤生長。然而,EGFR對全球m6A水平的調控尚不清楚。作者創建了國外m6A調節器簽名,包括寫入器(METTL3、METTL14、METTL16、VIRMA、RBM15、RBM15B、ZC3H13和WTAP)、擦除器(FTO和ALKBH5)和讀取器/調制器(YTHDF1、YTHDF2、YTHDF3、YTHDC1、YTHDC2、HNRNPC、IGF2BP3、CBLL1和HNRNPA2B1)。
為了直接研究EGFR在m6A調節中的作用,作者詢問了患者來源的GSCs的潛在功能關系。由于GSCs通常與EGF一起培養,作者將EGF從培養基中去除1周以避免偽像,然后用EGF配體處理兩個患者來源的間充質GSCs,以確定急性EGF配體處理對m6A水平的影響。選擇間充質GSCs是因為它們被認為更具侵襲性。使用點印跡法、比色法和免疫熒光法測量,EGF處理逐漸降低了m6A水平(圖1E-1G)。由于抗m6A抗體不完全具有特異性,作者采用液相色譜-質譜(LC-MS)并行分析,證實EGF處理降低了GSCs中m6A的表達(圖1H和1I)。在相互功能喪失研究中,作者用兩種臨床使用的EGFR抑制劑(厄洛替尼和拉帕替尼)治療GSCs。在沒有配體的情況下,EGFR可以在膠質母細胞瘤中被激活,包括通過組成型活性變體EGFRvIII的表達。總的來說,這些數據表明,與PDGFR誘導m6A相反,激活的EGF信號會下調GSCs中的m6A。
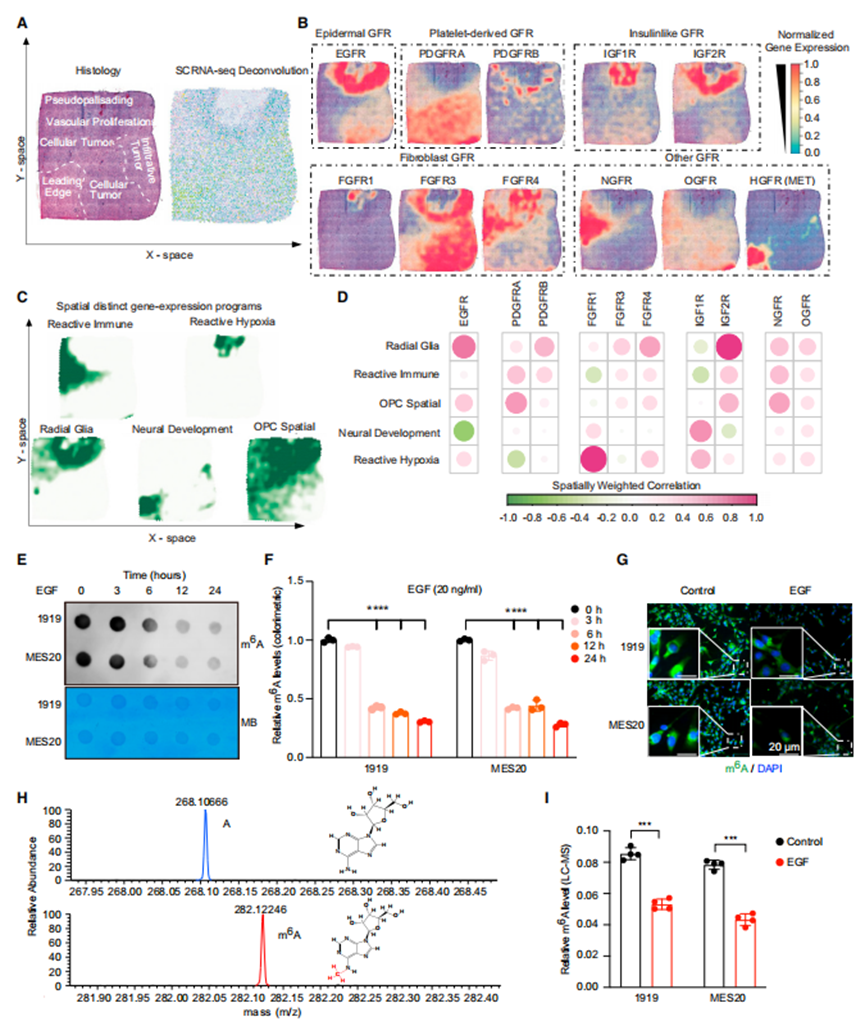 EGFR信號調控膠質母細胞瘤中RNA m6A水平
EGFR信號調控膠質母細胞瘤中RNA m6A水平
2、EGF促進ALKBH5的核定位
為了研究EGF信號調控m6A的機制,作者測量了EGF處理后GSCs中m6A寫入和擦除的總蛋白表達。EGF處理沒有改變GSCs中m6A調節因子的總蛋白水平(圖2A)。同樣,使用EGFR抑制劑厄洛替尼和拉帕替尼治療對m6A調節因子的蛋白水平沒有影響(圖2B)。由于m6A去甲基化酶FTO的細胞內分布決定了FTO與不同RNA底物的結合,作者測試了EGF是否改變了m6A調節因子的定位。EGF并未實質性改變GSCs中FTO或甲基轉移酶復合物(METTL3、METTL14和WTAP)的差異定位(圖2C)。相比之下,EGF處理誘導ALKBH5從細胞質轉移到細胞核(圖2C和2D)。在互失功能的研究中,用短發夾RNA(shRNA)靶向GSCs中的EGFR表達抑制ALKBH5的核定位(圖2E和2F)。在平行藥理學研究中,作者用EGFR抑制劑厄洛替尼處理GSCs,增加了細胞質ALKBH5的定位(圖2G)。組成活性EGFR EGFRvIII的表達促進了ALKBH5在GSCs中的核定位(圖2H)。其他ErbB家族成員(ERBB2/3/4)不直接結合EGF配體,但激活類似的細胞內通路,并可與EGFR形成異源二聚體。
接下來,作者推斷EGF誘導的ALKBH5核積聚在GSCs中可能是由于核輸入增加或向細胞質輸出減少。因此,作者測量了EGF處理對ALKBH5結合輸入蛋白(核轉運蛋白亞基α2, KPNA2;核轉運蛋白亞基β1, KPNB1)和輸出蛋白(染色體維持1 [CRM1])的影響。總的來說,EGFR活性促進ALKBH5從細胞核輸出減少,其功能是去除mRNA m6A修飾。
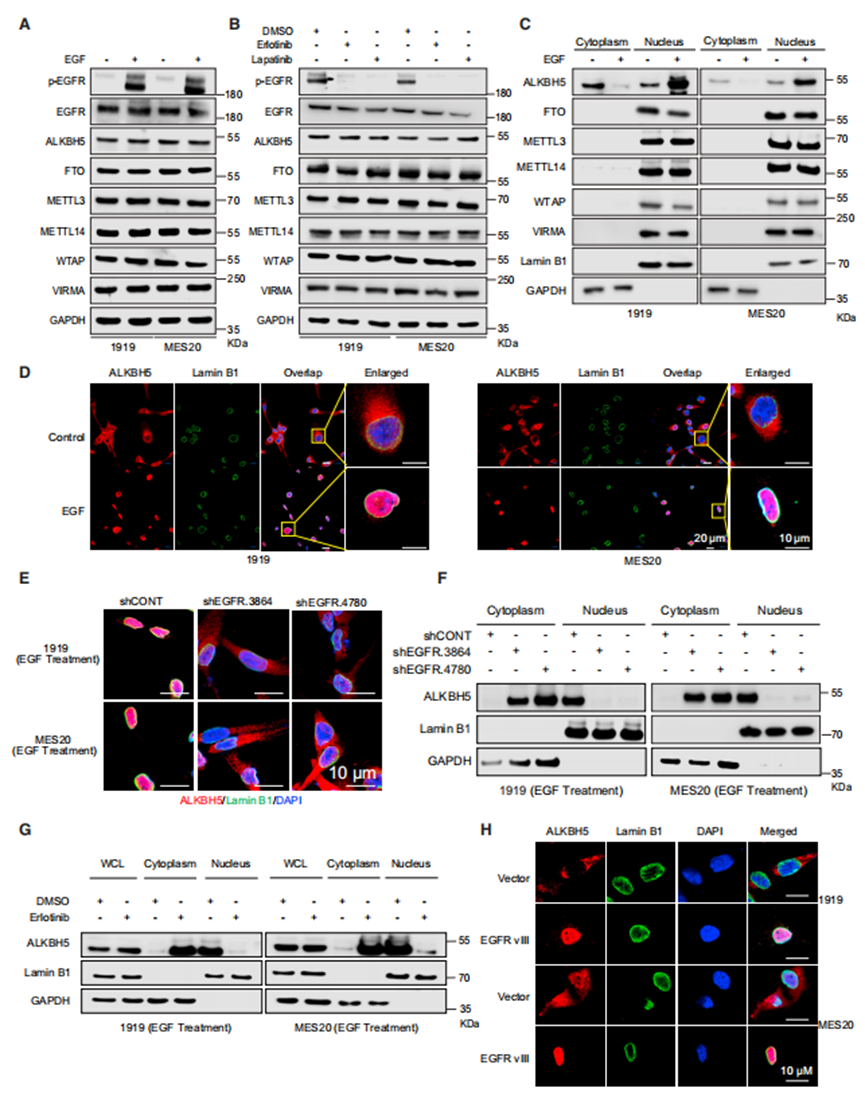 EGF-EGFR信號調控ALKBH5的核定位
EGF-EGFR信號調控ALKBH5的核定位
3、EGF誘導ALKBH5磷酸化,增加核定位和m6A去甲基化
ERK磷酸化METTL3以調節其功能。由于蛋白磷酸化可以調節許多分子的細胞內定位,作者接下來測試了EGFR是否通過ALKBH5磷酸化調節m6A水平和ALKBH5的細胞內定位。EGF處理誘導了GSCs中ALKBH5酪氨酸磷酸化,但沒有絲氨酸或蘇氨酸磷酸化(圖3A)。與EGF配體處理的作用平行,EGFR和EGFRvIII過表達誘導ALKBH5磷酸化(圖3B和3C)。反過來,EGFR抑制劑降低了GSCs中ALKBH5的磷酸化(圖3D)。
為了確定對EGF誘導的ALKBH5磷酸化重要的特定氨基酸殘基,作者使用GPS 5.0預測了ALKBH5蛋白內潛在的磷酸化位點,這表明71號氨基酸的酪氨酸是EGFR和SRC磷酸化的最高可能性位點(圖3E)。EGFR增加了野生型(WT) ALKBH5WT和ALKBH5Y306F的磷酸化,但沒有增加突變的ALKBH5Y71F的磷酸化(圖3F)。接下來,作者研究了ALKBH5Y71對ALKBH5細胞內定位和去甲基化酶活性的作用。用shRNA誘導GSCs減少內源性ALKBH5,然后用耐shRNA的ALKBH5進行轉導,EGF處理促進了ALKBH5WT的核定位,而不是ALKBH5Y71F的核定位(圖3G)。因此,Y71位點ALKBH5的磷酸化對于ALKBH5的核定位至關重要。
接下來,作者測試了ALKBH5磷酸化對m6A修飾和腫瘤細胞生長的影響。ALKBH5缺失的GSCs顯示出大量的m6A水平,而ALKBH5WT顯著降低了m6A水平,而ALKBH5Y71F則沒有(圖3H和3I)。m6A水平的調節反映在腫瘤細胞活力和體內腫瘤生長上。靶向ALKBH5的表達降低了體外細胞活力和體內腫瘤生長,通過重新表達ALKBH5WT而不是ALKBH5Y71F完全恢復了細胞活力(圖J - 3L)。總之,作者的數據表明,EGF誘導ALKBH5 Y71磷酸化,這對于ALKBH5核輸出和腫瘤細胞活力和體內生長至關重要。
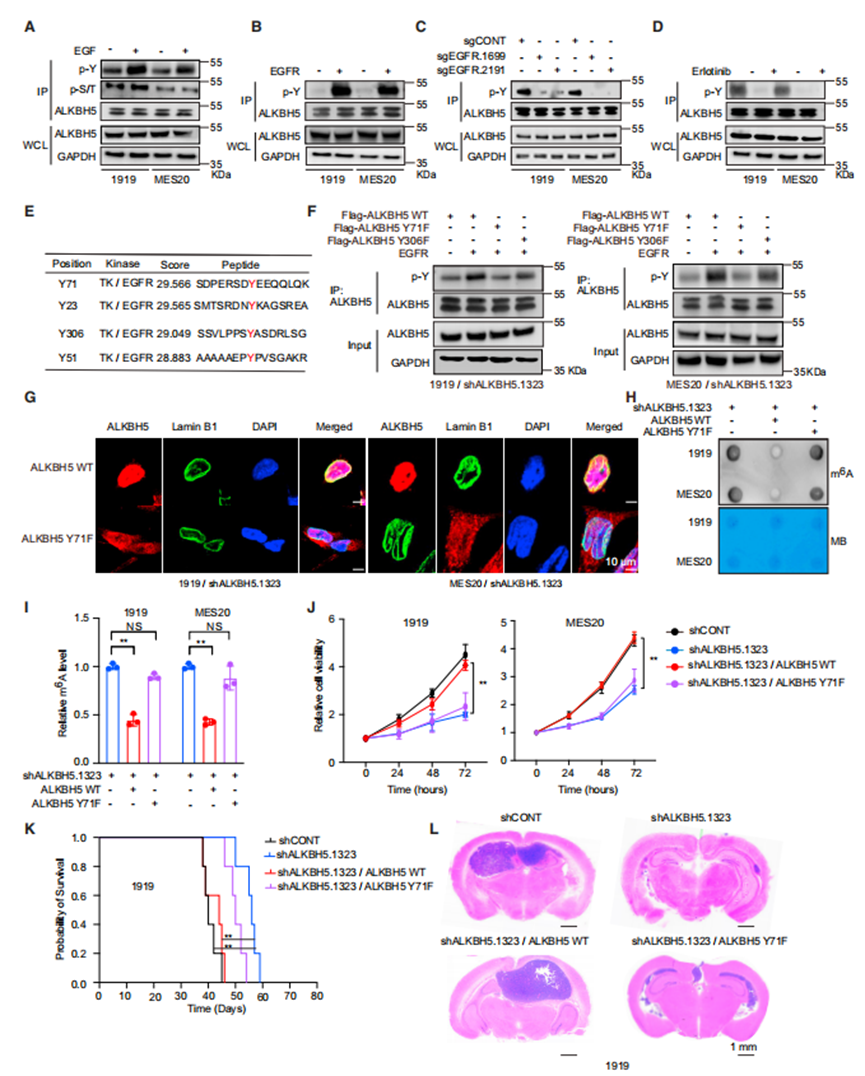 ALKBH5 Y71的磷酸化是其核定位及其在體內和體外功能的必要條件
ALKBH5 Y71的磷酸化是其核定位及其在體內和體外功能的必要條件
4、ALKBH5通過GCLM調控GSH合成
由于ALKBH5對GSC至關重要,作者尋找其作用的下游介質。Venn圖顯示了一個由17個基因組成的基因簇,其表達與ALKBH5相關,帶有m6A修飾,并且在GSCs和患者腫瘤中優先表達(圖4A)。在這些基因中,作者之前報道了GSCs對JMJD6和HEXB的依賴性。GSCs表達的GCLM蛋白水平高于NSCs(圖4B)、ACs(圖4C)和分化的膠質母細胞瘤細胞(DGCs,圖4D)。作者探討了該軸是否調節GSCs的干性。研究表明EGFR和ALKBH5而不是GCLM調節干性。
接下來,作者考慮了GCLM對膠質母細胞瘤生存能力和體內腫瘤生長的貢獻。作者用對照shRNA或兩個靶向GCLM的非重疊shRNA中的一個轉導了兩個患者衍生的GSCs。GCLM表達缺失降低了腫瘤細胞活力(圖4E)和荷瘤小鼠的存活率(圖4F)。
基于ALKBH5調節GCLM m6A修飾的假設,作者進行了甲基化(m6A)RNA免疫沉淀-定量聚合酶鏈反應(MeRIP qPCR),結果表明ALKBH5敲低增加了GCLM甲基化(圖4G)。支持ALKBH5磷酸化在下游靶標調控中的作用,ALKBH5缺失對GCLM m6A修飾的影響通過重新表達ALKBH5WT而不是ALKBH5Y71F完全恢復(圖4G),這表明ALKBH5的磷酸化對于GCLM m6A修飾至關重要。然后作者測試了m6A修飾對GCLM表達的作用。通過免疫印跡和免疫熒光檢測,在GSCs中靶向ALKBH5可以降低GCLM蛋白水平,通過重新表達ALKBH5WT而不是ALKBH5Y71F,可以完全恢復ALKBH5損失的影響(圖4H和4I)。作者探索了m6A修飾如何影響GCLM表達。ALKBH5敲低在兩個患者來源的GSCs中降低GCLM mRNA水平(圖4J)。由于m6A修飾經常導致m6A修飾mRNA的衰減,而ALKBH5的缺失增加了GCLM上m6A的水平,作者假設GCLM轉錄物的衰減可能受到ALKBH5的調節。事實上,用shAKBH5轉導的GSCs顯示出更快的GCLM轉錄物衰減(圖4K)。YTHDF2是一個m6A閱讀器,可以誘導mRNA衰變。GSCs的YTHDF2水平高于NSCs。因此,作者使用RBPsuite將YTHDF2結合映射到GCLM轉錄本上(圖4L)。為了測量YTHDF2與GCLM轉錄本的結合,作者進行了RNA免疫沉淀,然后使用IgG對照或YTHDF2抗體進行qPCR(RIP-qPCR),然后對GCLM進行qPCR。支持ALKBH5在調節YTHDF2結合中的作用,靶向ALKBH5表達的shRNA增強了YTHDF2與GCLM mRNA的結合(圖4M)。最后,作者測量了有YTHDF2藥理學抑制劑(DC-Y13-27)和沒有YTHDF2藥理學抑制劑(DC-Y13-27)時,SHAKBH5對GCLM mRNA水平的影響,發現抑制YTHDF2功能可以恢復ALKBH5調節后改變的GCLM水平(圖4N)。總的來說,作者的數據表明ALKBH5通過抑制YTHDF2介導的衰退來增加GCLM mRNA水平。
GCLM是GSH合成的限速步驟,因此作者測量了ALKBH5敲除或不敲除GSCs中的GSH水平。ALKBH5敲低降低了總GSH和還原型GSH水平,而ALKBH5WT而不是ALKBH5Y71F的重新表達完全恢復了ALKBH5缺失的影響(圖4O)。GCLM作為ALKBH5的下游介質發揮作用,GCLM的表達挽救了體內靶向ALKBH5的作用(圖4P和4Q)。ALKBH5共同調節GCLM m6A修飾和蛋白水平,以維持腫瘤生長。
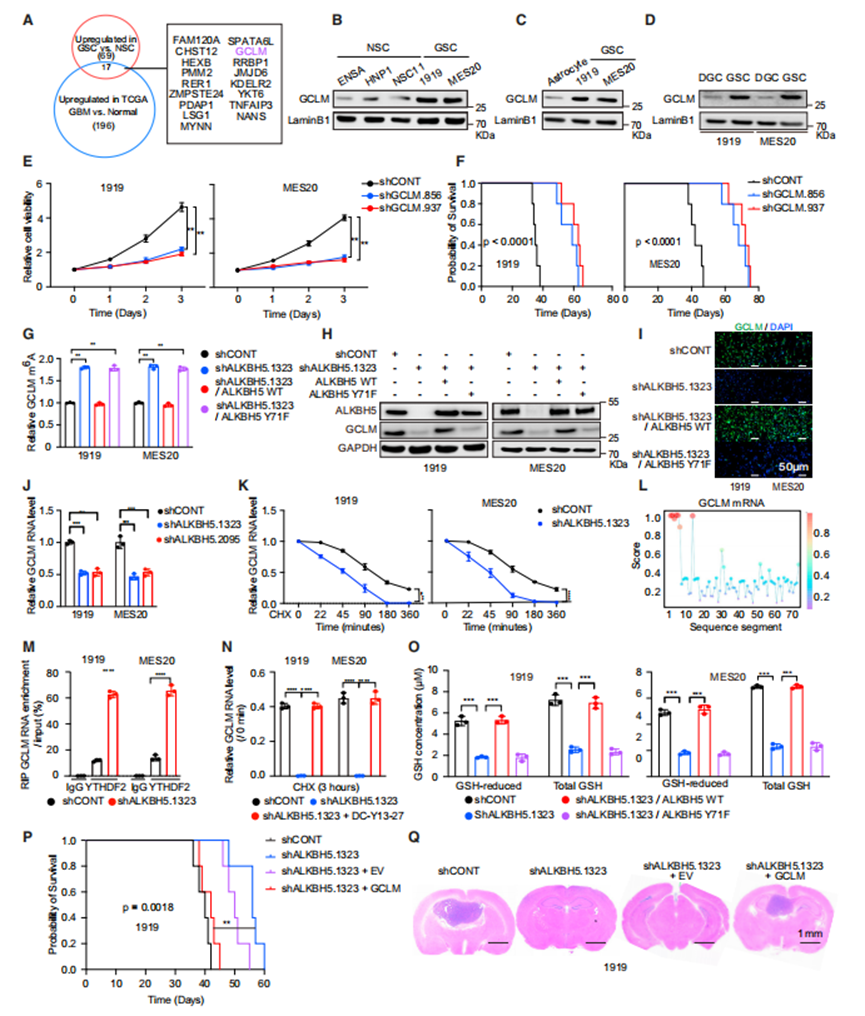 GCLM是ALKBH5在GSCs中的特異性靶點
GCLM是ALKBH5在GSCs中的特異性靶點
5、ALKBH5通過GCLM促進鐵死亡存活
鐵死亡是一種鐵依賴性的細胞死亡形式,由無限制的脂質過氧化和隨后的膜損傷引起。活性氧增加和脂質氧化是鐵死亡的兩個標志。先前的報道支持GSH在鐵死亡中的作用,以及GCLM在癌癥中鐵死亡的調節中的作用。GCLM敲低誘導GSC鐵死亡形態和細胞死亡(圖5A-5C)。電鏡顯示,GCLM在GSCs中敲低導致線粒體大小減小,線粒體膜密度增加,線粒體脊丟失(圖5D)。靶向ALKBH5表達表型復制了在電子顯微鏡下觀察到的GSC結構改變和誘導細胞死亡(圖5E和5F)。為了證明ALKBH5通過調節GSH誘導鐵死亡,作者將ALKBH5的表達定位在GSCs中,然后通過提供還原型GSH來測量其拯救作用的能力。ALKBH5敲低降低了細胞活力,增加了SYTOX+細胞群,通過減少GSH完全逆轉了這一點(圖5G和5H)。靶向ALKBH5表達可誘導ROS和氧化脂質的積累,而通過降低GSH可逆轉這一過程(圖5I-5L)。因此,EGFR-ALKBH5-GCLM通過GSH調節來保護鐵死亡細胞。
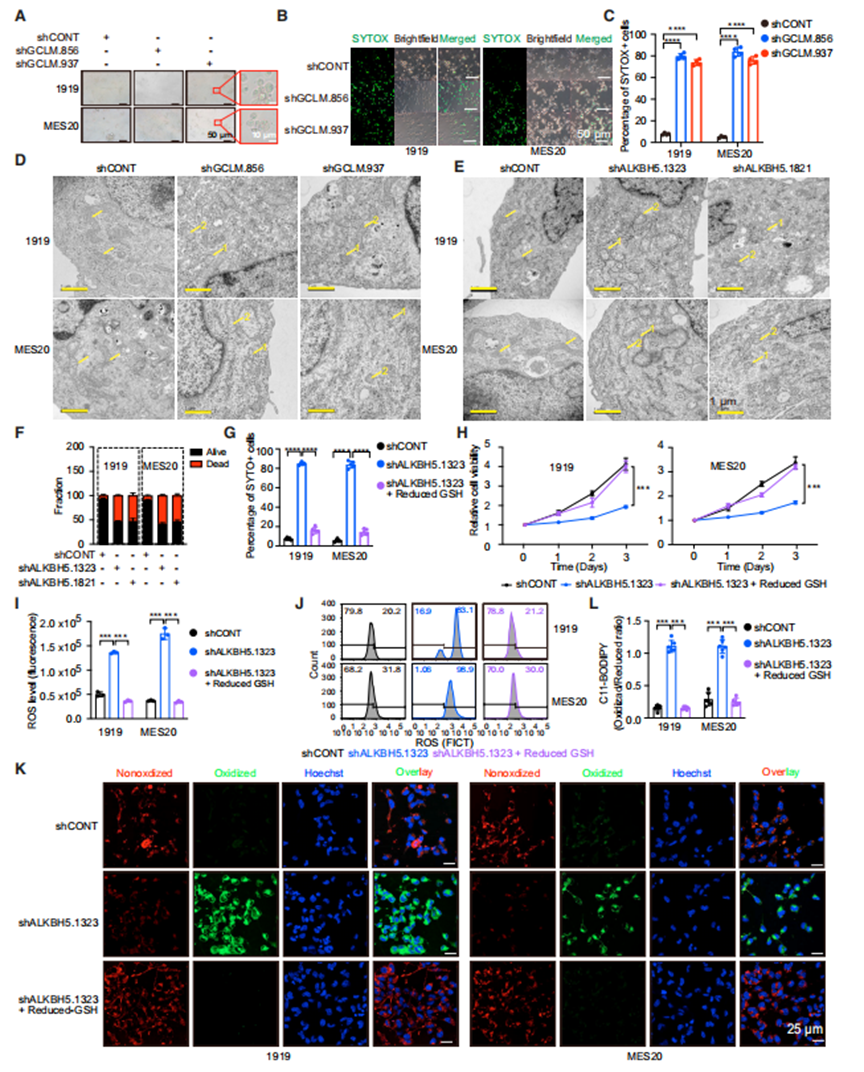 ALKBH5通過GCLM抑制鐵死亡
ALKBH5通過GCLM抑制鐵死亡
6、ALKBH5的藥理學靶向增強了EGFR抑制的抗腫瘤效果
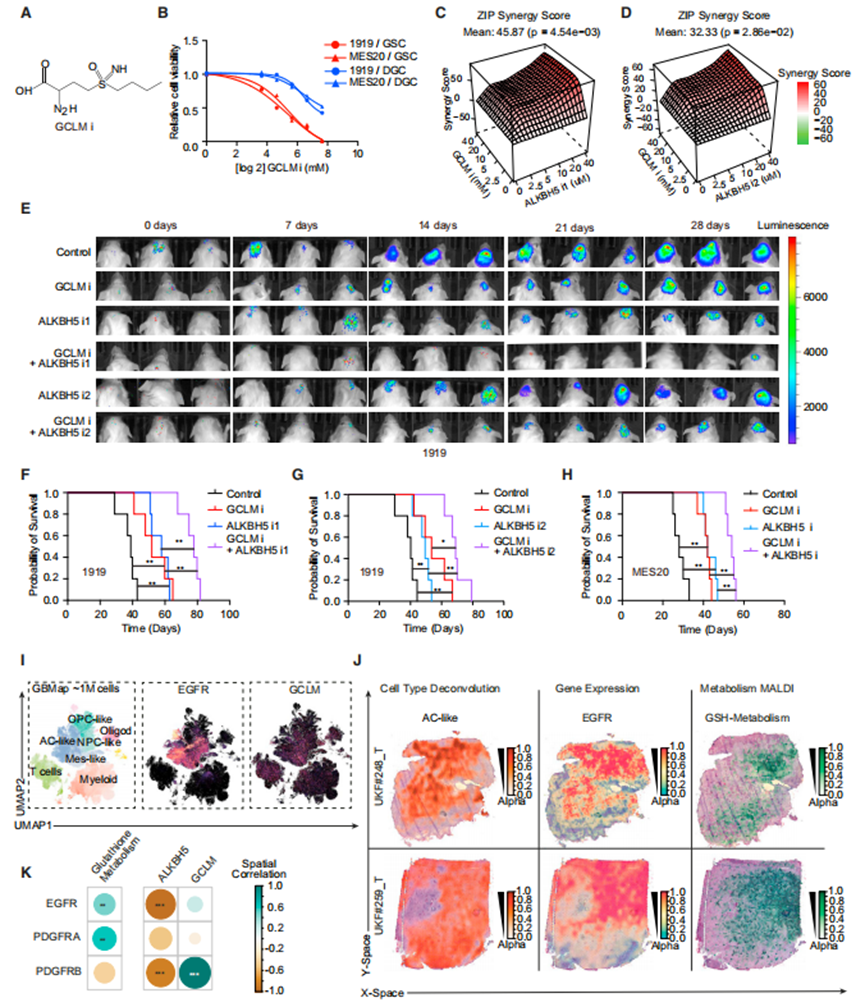
作者選擇了美國食品和藥物管理局(FDA)批準的EGFR抑制劑(erlotinib)和兩種抑制ALKBH5酶活性的化合物(指定為ALKBH5抑制劑1 [ALKBH5i1]和ALKBH5抑制劑2 [ALKBH5i2])(圖6A)。使用ALKBH5抑制劑治療可增加GSCs中的m6A水平,支持ALKBH5抑制劑的靶效應(圖6B)。基于EGFR和ALKBH5之間的分子相互作用,作者假設聯合這些抑制劑可以提供更大的抗腫瘤效果。ALKBH5抑制劑增強了厄洛替尼對GSCs的療效(圖6C)。
無論是使用ALKBH5i還是厄洛替尼,其療效與單一藥物相似,都能減少腫瘤在體內的生長,而聯合治療能更大程度地減少腫瘤體積(圖6D)。每一種藥物單獨治療時,腫瘤生長的減少轉化為原位荷瘤小鼠的生存期延長,聯合治療時生存率提高(圖6E-6G)。為了成為一種可行的腦腫瘤治療藥物,藥物需要有一個治療指數和進入中樞神經系統,因此作者評估了這些藥物聯合在體內的毒性和分布。用EGFR和ALKBH5抑制劑單獨治療或聯合治療均未引起肝毒性的實驗室跡象,通過血清天冬氨酸轉氨酶和丙氨酸轉氨酶活性水平測量(圖6H和6I),也未引起肝、腎或心臟損傷的組織學跡象(圖6J)。為了測量藥物傳遞,作者用EGFR或ALKBH5抑制劑治療患有膠質母細胞瘤的原位異種移植物小鼠,然后作者收集血清、腫瘤和非腫瘤腦來測量藥物水平。
接下來,作者基于EGFR和ALKBH5的表達,比較了TCGA和中國膠質瘤基因組圖譜(CGGA)中膠質母細胞瘤患者的生存率。腫瘤中EGFR和ALKBH5均高表達的患者預后較低表達的患者差(圖6K和6L)。綜上所述,ALKBH5與EGFR聯合治療膠質母細胞瘤患者是一個潛在的治療靶點。
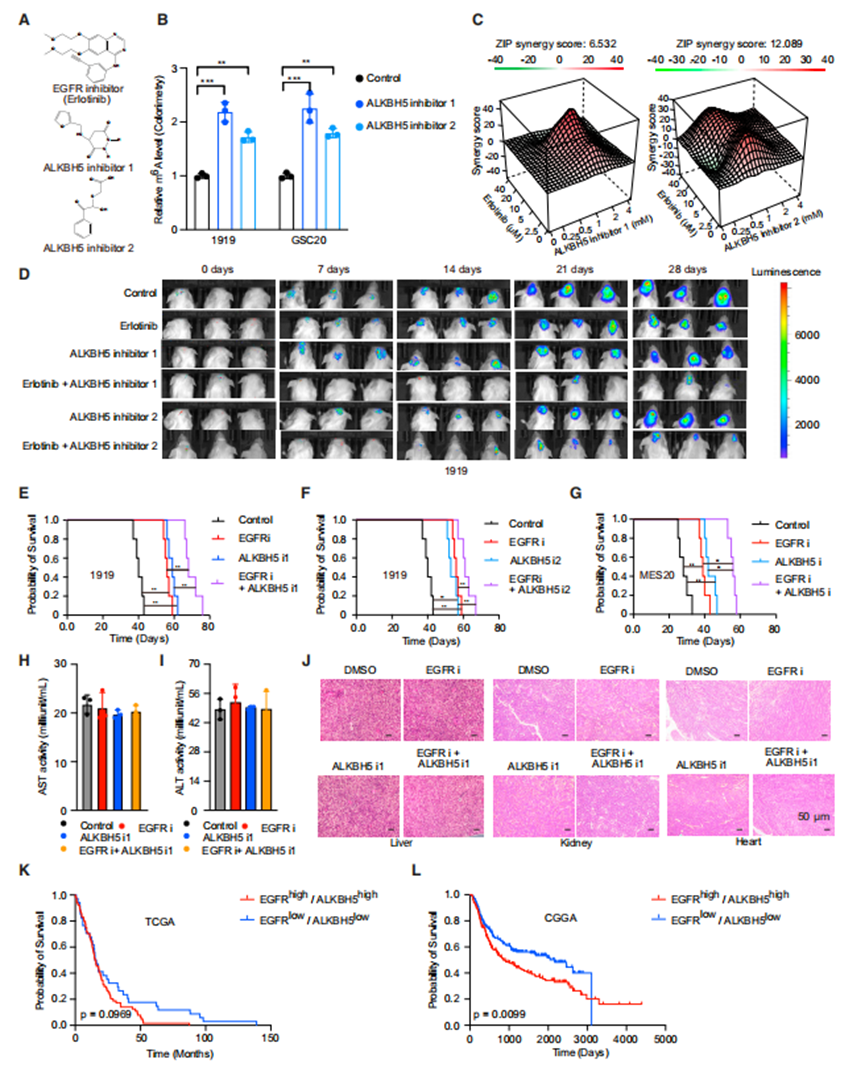 ALKBH5的藥理靶向增強了EGFR的抗腫瘤作用
ALKBH5的藥理靶向增強了EGFR的抗腫瘤作用
7、ALKBH5的藥理學靶向增強了GCLM抑制劑的抗腫瘤功效
誘導鐵死亡的藥物已被描述為潛在的輔助抗癌治療。癌細胞具有更高的鐵代謝需求,使其比正常細胞更容易發生鐵死亡,而GSCs優先運輸鐵。BSO是一種有效的、特異性的、選擇性的不可逆GCLM抑制劑(圖7A)。BSO誘導鐵死亡,并表現出相對于DGCs對GSCs的優先活性(圖7B)。與EGFR和ALKBH5一樣,ALKBH5和GCLM似乎是順序連接的,但大多數分子節點具有多個輸入和輸出,包括反饋機制。這導致了垂直整合治療的組合方法,特別是b-raf原癌基因,絲氨酸/蘇氨酸激酶(BRAF)和絲裂原活化蛋白激酶(MAPKK, MEK)抑制劑在黑色素瘤中的應用。作者假設聯合靶向ALKBH5和GCLM可能會顯示出額外的益處。事實上,ALKBH5和GCLM抑制劑在體外聯合使用顯示出協同抗GSC的功效(圖7C和7D)。用生物發光標記轉導的膠質母細胞瘤原位異種移植物小鼠接受了載體對照、ALKBH5抑制劑、GCLM抑制劑或聯合治療。單獨使用ALKBH5i或GCLM抑制劑治療可減少體內腫瘤體積,并具有體內聯合治療的額外益處(圖7E)。通過ALKBH5和GCLM抑制劑的聯合治療獲益,減少腫瘤體積與延長生存期有關(圖7F-7H)。
最后,作者考慮了ALKBH5-GCLM、EGFR-GCLM和EGFR-ALKBH5-GCLM的預后意義。為了在沒有細胞培養的情況下證實EGFR和GCLM之間的聯系,作者詢問了膠質母細胞瘤患者標本的空間轉錄組學和代謝組學數據。EGFR和GCLM mRNA在單細胞水平上相關(圖7I),EGFR mRNA水平在空間上與GSH代謝相關(圖7J和7K),盡管不是完全相關。綜上所述,靶向表觀轉錄組調控和鐵死亡聯合應用代表了膠質母細胞瘤的潛在治療模式。
 以GCLM為靶點,誘導鐵死亡,對GSCs產生抗腫瘤作用
以GCLM為靶點,誘導鐵死亡,對GSCs產生抗腫瘤作用
結論:
膠質母細胞瘤在微環境因素和治療下調節RTKs的下游效應物,這表明同時靶向這些信號通路的多種成分對于避免耐藥至關重要。作者的研究結果表明,聯合靶向EGFR和ALKBH5是有希望的,厄洛替尼和ALKBH5抑制劑在體外和體內都有聯合益處。對鐵死亡的敏感性取決于參與ROS、鐵、脂質和能量代謝的基因和途徑。盡管GSC對傳統的鐵死亡抑制劑具有耐藥性,但GCLM抑制劑選擇性地抑制GSC的生長和腫瘤形成。GCLM和ALKBH5抑制劑對GSC生長有協同作用。這些結果表明,靶向該通路中的多個節點可能是有益的,可能表明存在正反饋回路或多重相互作用,需要中斷以獲得最佳治療效果。總的來說,這些藥物組合值得進一步研究。
實驗方法:
GSC提取;細胞培養;質粒分離和定點誘變;逆轉錄病毒包裝與感染;體內腫瘤發生;患者數據庫和生物信息學;細胞活性;細胞分級分離;免疫印跡;免疫熒光分析;免疫共沉淀;mRNA純化;m6A斑點雜交;m6A定量分析;m6A RNA修飾的LC-MS定量分析;LC-MS/MS分析藥物濃度;β-半乳糖苷酶細胞染色;ALT活性檢測;AST活性檢測;MeRIP-qPCR;活性氧檢測。
參考文獻:
Lv D, Zhong C, Dixit D, Yang K, Wu Q, Godugu B, Prager BC, Zhao G, Wang X, Xie Q, Bao S, He C, Heiland DH, Rosenfeld MG, Rich JN. EGFR promotes ALKBH5 nuclear retention to attenuate N6-methyladenosine and protect against ferroptosis in glioblastoma. Mol Cell. 2023 Nov 8:S1097-2765(23)00862-6. doi: 10.1016/j.molcel.2023.10.025. Epub ahead of print. PMID: 37979586.