






教你四張圖搞定一篇nature communication

編碼轉錄因子(TF)基因的致病突變可以影響TF與其同源DNA結合基序的相互作用。TF突變是否以及如何影響與TF復合元件(CE)的結合以及與其他TF的相互作用尚不清楚。在這里,作者報道了B細胞特異性紊亂的人類淋巴瘤中,特別是經典的霍奇金淋巴瘤中TF改變的獨特機制。它是由一種復發性體細胞錯義突變c.295 T>c(p.Cys99Arg;p.C99R)引起的,該突變靶向干擾素調節因子4(IRF4)的DNA結合域中心,IRF4是免疫細胞中一種關鍵TF。IRF4-C99R從根本上改變了IRF4-DNA的結合,失去了與經典IRF基序的結合,并獲得了與經典和非經典IRF CE結合的新形態。IRF4-C99R通過阻斷IRF4依賴性漿細胞誘導徹底改變IRF4功能,并以非經典激活蛋白-1(AP-1)-IRF-CE(AICE)依賴性方式上調疾病特異性基因。這些數據解釋了單個突變如何導致TF特異性和基因調控的復雜轉換,并為特異性阻斷突變TF的新形態DNA結合活性開辟了前景。該研究于2023年11月發表在《nature communication》,IF 16.6分。
技術路線
主要研究結果
1. IRF4-C99R突變在人類淋巴瘤中復發
通過挖掘和整合cHL細胞系的基因組和轉錄組數據,作者在7個HL細胞系中的2個細胞系,即Bcell衍生的HRS細胞系L428和U-HO1中,鑒定并驗證了IRF4基因中相同的c.295 T>c(chr6:394899 T>c;hg38)突變體(圖1a)。基于ANNOVAR整合的各種計算機分析(包括SIFT、Polyphen2、MutationTaster、FATHMM、CADD評分),一致預測該變體是有害的。此外,除了在AA100中攜帶錯義突變的單一等位基因(1/251478)外,在影響相鄰AA90-104的gnomAD中沒有發現種系非同義單核苷酸變體。在HL細胞系中,c.295 T>c突變等位基因伴有至少一個野生型(WT)IRF4基因拷貝,并且WT和突變IRF4 mRNA轉錄物都同等被檢測到(圖1a)。由于HRS細胞在受影響的淋巴結中很少見,通過激光顯微切割HRS細胞的DNA-PCR驗證了IRF4 c.295 T>c在20個原發性cHL樣本中的4個樣本中存在。IRF4 c.295 T>c最近在原發性縱隔B細胞淋巴瘤(PMBCL)中被描述,該淋巴瘤實體與cHL具有不同的生物學特征。從486例PMBCL病例的不相關隊列中平行挖掘靶向基因組測序數據,在486例病例中的29例(5.9%)中發現了相同的IRF4 c.295 T>c突變。相反,IRF4 c.295 T>c在其他淋巴瘤類型中很少被記錄,如DLBCL。此外,C99R c.295 T>c(chr6:394899 T>c;hg38)突變的基因組位置在外顯子3內,因此位于轉錄起始位點(TIS)下游>3kb。此外,它缺乏典型的熱點RGYW基序,表明這種突變不是由B細胞淋巴瘤的異常體細胞超突變引起的,該突變通常影響TIS下游約2-2.5kb的區域。
IRF4在末端B細胞分化階段控制漿細胞基因表達程序,盡管在所有亞型中都有高水平的IRF4表達,但在HRS細胞中基本缺乏該程序。在IRF4 c.295 T>c突變中,堿性AA精氨酸取代AA 99位置的中性AA半胱氨酸(Cys;c)(p.Cys99Arg;C99R),其在從人類到斑馬魚的IRF4中高度保守,也在大多數其他IRF家族蛋白的DBD中高度保守(圖1a)。C99R位于IRF4 DBD的α3-識別螺旋的中心,并緊鄰Arg98,Arg98對特異性IRF4 DNA結合至關重要。這一發現表明C99R可能干擾IRF4:DNA復合物的形成,從而干擾IRF4的轉錄活性。
2. IRF4-C99R功能喪失ISRE,但被功能激活
為表征IRF4-C99R,首先探索了其與干擾素刺激反應元件(ISRE)的DNA結合活性,該元件包含三個共有基序5’-GAA-3’(圖1b),這是IRFs識別的關鍵基序之一。與IRF4-WT不同,IRF4-C99R不與ISRE結合,如電泳遷移率偏移測定(EMSA)所示。然而,IRF4-C99R突變的復發性和在cHL中的高水平表達表明,這種突變可能不僅構成功能喪失的異常,而且可能具有額外的從頭開始的功能。為了分析IRF4-C99R功能,作者產生了四環素(Tet)誘導的表達IRF4-C99和IRF4-WT的BJAB B細胞非霍奇金淋巴瘤細胞的大量培養,其僅在低水平下表達內源性IRF4。時間進程基因表達分析顯示,與IRF4-WT相比,IRF4-C99R改變了一組特殊但較少基因的表達(圖1c)。這與其失去的結合經典ISRE基序的能力一致。IRF4-C99R與IRF4-WT一樣有效地挽救了HRS細胞,使其免于由shRNA介導的內源性IRF4敲除誘導的細胞死亡,從而證實了其功能。
3. IRF4-C99R從根本上改變了IRF4的DNA結合特異性
與在ISRE DNA基序上形成低親和力同源二聚體或多聚體復合物相反,有效的IRF4 DNA結合需要不同的伴侶,如CEs25-27的ETS和AP-1蛋白。鑒于cHL中普遍不存在ETS TF,作者認為IRF4與HRS細胞中EICE的結合是不可能的。然而,具有高水平JUNB和BATF表達的組成型AP-1活性是HRS細胞的標志。因此,作者推測IRF4-C99R通過DNA結合與最近鑒定的AICE來調節基因表達,間距為4bp 的5’-IRF(TTTC)/nnnn/AP-1(TGASTCA)-3’ (AICE1),或無間距的5’-IFF(GAAA)/AP-1(TGASTCA-3’(AICE2),它們都調節免疫細胞中的關鍵轉錄程序。為評估這一假設,作者監測了在強親和力(標記為“AICE1(Ctla4)”)、弱親和力(AICE1(IL12Rb))或中間親和力(AICE2(Bcl11b))AICE的基序條件下IRF4-JUNB/BATF-DNA復合物的形成(圖1d)。雖然觀察到AICE1(Ctla4)和AICE1的IRF4-C99R結合完全喪失(稱為AICE結合模式1(BP1)),但與IRF4-WT(BP2)相比,AICE2(Bcl11b)的IRF4-C99R結合增強(圖1d)。IRF4-C99RS104T的行為與IRF4-C992R相似(圖1b、d)。引人注目的是,從5’-GAAA-3’到5’-TTTC-3’的AICE2(Bcl11b)中IRF基序的反向互補(稱為AICE2FLIP)揭示了僅突變體IRF4-C99R-JUNB/BATF-DNA復合物的形成(圖1e,AICE2FLIP,BP3)。此外,AICE復合物的形成通常需要相對于AICE2位于?4 bp(-4T)的胸腺嘧啶(稱為AICE2-4T)。IRF4-C99R克服了這種限制,因為它在不存在-4T的情況下形成強的DNA結合復合物,這導致IRF4-WT結合的損失(圖1f;AICE2-4C;BP4)。類似地,與IRF4-WT相比,與c-JUN(JUN)/BATF異二聚體AP-1復合物一起觀察到IRF4-C99R結合模式的改變。
此外,進行結構建模分析以提供關于IRF4-C99R突變如何影響與ISRE和AICE1 DNA結合基序的相互作用(圖1g)。對于結構模型,IRF4和DNA的初始結構是從以前的晶體結構(PDB:7JM4)中獲得,并且基于所得的對接參數(如HADDOCK評分、簇大小和去溶劑化能)來考慮最可行的模型。如圖1g所示,對于IRF4:ISRE相互作用,用Arg直接取代C99導致與DNA堿基的空間沖突。為了適應C99R和ISRE之間的相互作用,dsDNA必須彎曲和/或扭結。對于IRF4-C99R:AICE1結合,較差的對接得分表明,如較差的能量最小化結構所反映的那樣,極不可能發生顯著的相互作用。因此,盡管AICE1建模可能由于DNA畸變而受到限制,但它表明IRF4-C99R不與AICE1結合,這與作者的EMSA結果一致。
在僅包含IRF4(AA 20–139)、JUNB(AA 269–329)和BATF(AA 28–87)的DBD重組蛋白中也觀察到了IRF4-C99R DNA結合特性的這些改變模式。此外,通過單分子熒光顯微鏡和交錯延時照明觀察了IRF4-C99R或IRF4-WT的DNA結合片段, IRF4-C99R的長結合DNA連接(>2s)的百分比與IRF4-WT相當。
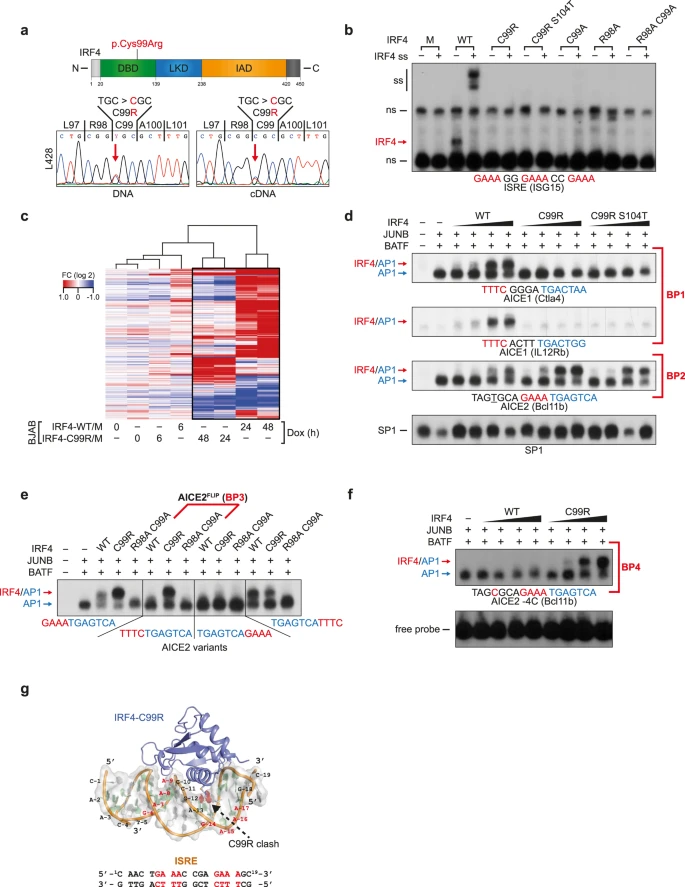
圖1:IRF4-C99R功能表征和基本的DNA結合改變
4. IRF4-C99R淋巴瘤細胞中IRF4 DNA結合模式和協同活性發生全局改變
接下來,為詳細特異性地繪制HRS細胞中開放性染色質,首先從HRS細胞系L428(攜帶IRF4-C99R和KM-H2,表達IRF4-WT)以及作為對照的非霍奇金、非IRF4表達REH細胞中生成高分辨率全基因組DNA酶I超敏位點(DHS)和數字足跡數據。DNaseI切割頻率的分析揭示了對DNaseI消化的保護作用,表明蛋白質復合物的占據,以及僅在HRS細胞中AICE2(BP2)、AICE2FLIP(BP3)、AICE2?4T和AICE2-4C(BP4)側翼區域的可及性提高(圖2a)。值得注意的是,這些基序在L428IRF4-C99R中富集和保護程度最高(圖2a)。這些AICE2基序在L428IRF4-C99R細胞中的共定位分析揭示了與AP-1基序共定位的突變體特異性位點相對應的特異性簇,但與通常參與B細胞和HL細胞基因調控的其他TF位點不一致。這些發現再次支持IRF4-C99R賦予了細胞不同的表達譜的。
為定義L428或KM-H2-特異性DHS組,作者研究了L428IRF4-C99R和KM-H2IRF4-WT細胞之間的標簽計數比,并根據它們在DNaseI-seq信號中的倍數變化對它們進行排序(圖2b)。L428IRF4-C99R-特異性DHS(即第3組)與這些細胞中上調的基因表達相關(圖2b)。然后確定了AICE2(BP2)、AICE2FLIP(BP3)、AP-1和ISRE基序在不同DHS組中的富集情況。L428IRF4-C99R-特異性DHS富集AICE2、AICE2FLIP和AP-1基序,但缺失ISRE基序,而KM-H2IRF4-WT特異性DHs缺失AICE2,AICE2FLIP和AP--1基序,卻富集ISRE(圖2c)。使用HOMER對細胞系特異性DHS中TF基序的搜索結果顯示,AICE2和AICE2FLIP是L428IRF4-C99R-特異性DHSs中富集度最高的兩個基序,而不是KM-H2IRF4-WT-特異性DHSs位點(圖2d)。相反,ISRE基序在KMH2IRF4-WT-而不是L428IRF4-C99R-特異性DHS中富集,這再次表明IRF4-C9R轉移結合位點至AICE2基序(圖2d)。L428IRF4-C99R與KM-H2IRF4-WT細胞DHSs的基因集富集分析(GSEA)顯示,在上調的基因中,AICE2基序足跡增加,論證了該基序在AICE2 L428IRF4-C99R細胞中的功能相關性。
最后,作者在L428IRF4-C99R和KM-H2IRF4-WT細胞中進行了全基因組范圍的JUNB和IRF4 chip-seq分析(圖2e-i)。IRF4-JUNB-ChIP峰內的序列緊密聚集(圖2e),并且與KM-H2IRF4-WT和GM12878細胞相比,L428IRF4-C99R細胞中的序列顯示出更大的重疊(圖2f),這與IRF4-C99 R與IRF和AP-1 CE的強制結合一致。盡管與GM12878相比,兩個HRS細胞系中的IRF4 ChIP峰值頻率更高(圖2f),但在KM-H2IRF4-WT細胞中與JUNB的重疊要低得多。當單獨排序時,IRF4和JUNB在L428IRF4-C99R中顯示出高度相似的結合模式,但在KM-H2IRF4-WT細胞中沒有,這對應于開放的染色質區域,并與基因表達增加有關(圖2g)。與這些分析和DHS數據集的基序發現結果一致(圖2h),在L428IRF4-C99R-特異性IRF4-CIP峰中,AICE2(BP2)和AICE2FLIP(BP3)基序頻率增加,相反地,與KM-H2IRF4-WT特異性ChIP峰相比,ISRE基序頻率較低。當將L428的IRF4和JUNB染色質結合模式與GM12878細胞進行比較時,也觀察到了這些發現。重要的是,GSEA揭示了IRF4和JUNB-CIP峰與L428IRF4-C99R細胞中基因表達增加有關,但與KM-H2IRF4-WT細胞中的基因表達無關。
在ChIP-seq數據集中發現AICE1(BP1)是KM-H2IRF4-WT細胞中最重要的基序,但在L428IRF4-C99R細胞中未被鑒定到(圖2i)。AICE2(BP2)出現在兩個數據集中最重要的基序中,在L428IRF4-C99R細胞中更為重要,其中共鑒定了五種基序類型(圖2i)。分析還揭示了AICE2FLIP(BP3)在L428IRF4-C99R細胞中的獨特重要性。這些結果與DNA結合研究一致(圖1),并進一步支持IRF4-C99R從根本上改變了淋巴瘤細胞中IRF4全基因組DNA結合模式,并在不同的新AICE上加強與AP-1/JUN TF協同結合。
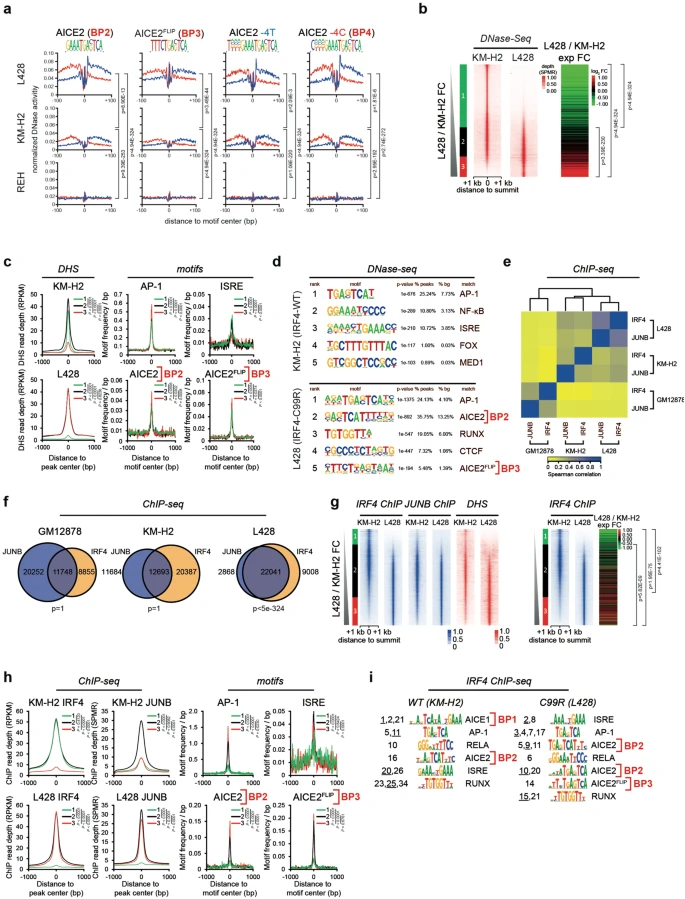
圖2:IRF4-C99R與C99R突變陽性淋巴瘤細胞中規范和非規范AICE2位點的全基因組增加和不同的DNA結合模式有關
5. IRF4-C99R破壞原代B細胞中IRF4的功能并重新編程基因表達
為進一步探索IRF4-C99R在B細胞中表達的功能,用IRF4-WT、IRF4-C998R或功能喪失(LOF)突變體IRF4-R98AC99A作為對照,逆轉錄病毒轉導原代小鼠C57BL/6脾臟B細胞(圖3a)。在已知的IRF4-R98AC99A LOF突變體中,與IRF4:DNA復合物形成密切相關的殘基R98和C99都被丙氨酸(A)取代,從而消除IRF(4)的DNA結合和功能。用LPS和IL?4培養的B細胞導致強大的內源性IRF4表達,并誘導約30%的漿母細胞,其特征是CD138高和B220低表型。在非功能性IRF4-R98AC99A變體異位表達后獲得相同的結果(圖3a)。IRF4-WT異位表達后,約70%的細胞轉化為漿母細胞表型。相反,IRF4-C99R減少了發育中的漿細胞數量,即阻斷了固有漿細胞形成,這表明IRF4-C99在B細胞終末分化方面具有主要的負向調節功能(圖3a)。為檢測基因表達的變化,作者分離了用不同IRF4變體轉導的小鼠C57BL/6脾臟B細胞,然后進行RNA-seq分析(圖3b–f)。總體而言, IRF4-C99R顯示出與R98AC99A LOF變體相比更類似于IRF4-WT的轉錄譜(圖第3b段)。IRF4-C99R調節減少的基因集(圖3c),包括IRF4-WT靶基因表達的廣泛損失以及新靶點的增加(圖3d)。修飾的脾 B 細胞的 mRNA 表達譜與來自各種造血細胞類型的 mRNA 表達譜的集成顯示 IRF4-C99R 調節整體 IRF4-WT 誘導和漿細胞特異性基因表達的阻斷(圖 3e),證實 IRF4-C99R 無法指導 IRF4 定向漿細胞進程。同時,IRF4-C99R 上調髓系相關基因(圖 3e、f)。總之,這些數據證實了與IRF4-WT相比,IRF4-C99R依賴性基因調控和功能的根本變化。
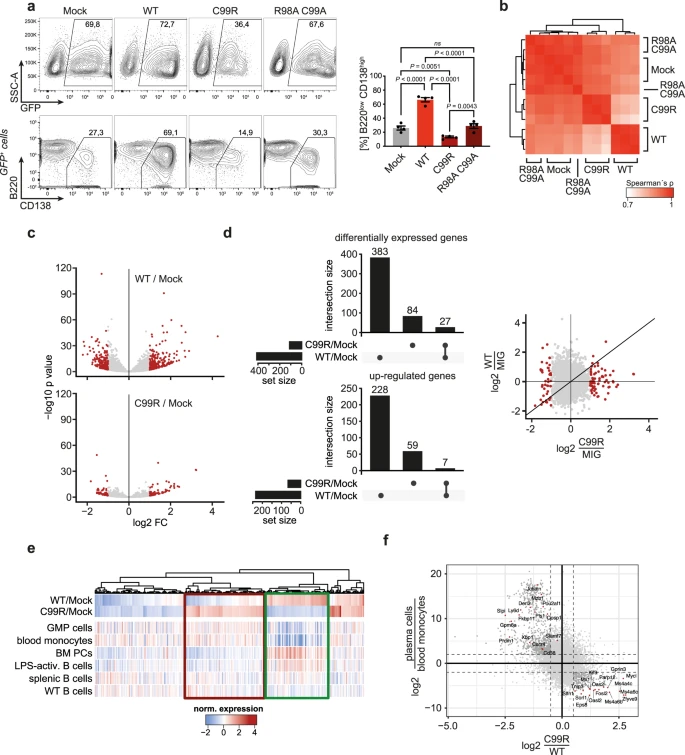
圖3:與IRF4-WT相比,IRF4-C99R阻斷IRF4依賴性漿細胞誘導并調節數量較少但不同的基因
6. IRF4-C99R通過非經典AICE激活淋巴瘤特異性基因表達
為將IRF4-C99R調節的基因與cHL的HRS細胞特異性固有基因直接聯系起來,作者整合了來自脾臟B細胞的RNA-seq數據與HRS細胞特異性基因表達譜(圖4a)。在 IRF4-C99R 僅上調而非 IRF4-WT 上調的 HRS 細胞特異性表達基因中,GATA3、CCL5(也稱為 RANTES)和 TNFRSF8 (CD30),這三種基因都是最突出的 cHL 標志性基因,以及 CD80、PDE4D 和 CASP6(圖 4a)。
為了進一步剖析這些基因的IRF4-C99R特異性誘導機制,重新分析了L428IRF4-C99R細胞特異性IRF4-JUNB ChIP峰的ChIP-Seq數據,但在KMH2IRF4-WT細胞中未發現。作者專注于調控GATA3表達的區域,在L428IRF4-C99R特異性IRF4-JUNB ChIP峰中鑒定了幾個AICE2樣CE,分別為GATA3Peak_1(5’-TGAGTCAGAGA-3’)、GATA3Peak_2(5’-TAAATGAGTCA-3’)和GATA3Peak_3(5’-GGAATGAGTCA-3’)(圖4b,左)。DNA結合研究表明,IRF4-C99R在這些位點形成IRF-AP-1復合復合物,而IRF4-WT不與這些串行結合(圖4b,右,AP-1由JUNB/BATF異二聚體組成)。值得注意的是,這些位點均不包含經典的5’-GAAA-3’IRF基序,而是包含其非經典簡并變體。這些結果表明,與C99相比,IRF4 R99在這些基序上的靈活性更高,類似于對圖1e-g中描述的AICE2變體的觀察。與KM-H2IRF4-WT細胞相比,在使用ExplaiNN的ChIP-seq數據中鑒定的含有IRF基序在L428IRF4-C99R細胞中退化得更多,這反映了IRF4-C99 R對退化的半含ISRE基序的結合能力的增加(圖 4c),這在 AICE 基序中最為明顯(圖 4c)。通過分析熒光素酶報告基因構建體證實了 IRF4-C99R 介導的 GATA3Peak_1 組件的轉錄活性(圖 4d)。在這里,IRF4-C99R 與 AP-1 TFs JUNB 和 BATF 聯合使用特異性增強了熒光素酶活性,而 IRF4-WT 則沒有。最后,攜帶IRF4-C99R突變的HRS細胞系與缺乏IRF4-C99 R的HRS的表達譜與cHL特異性基因關系的比較表明,cHL標志性基因GATA3、CCL5和TNFRSF8的表達在IRF4-C99R細胞系中特別高(圖4e)。
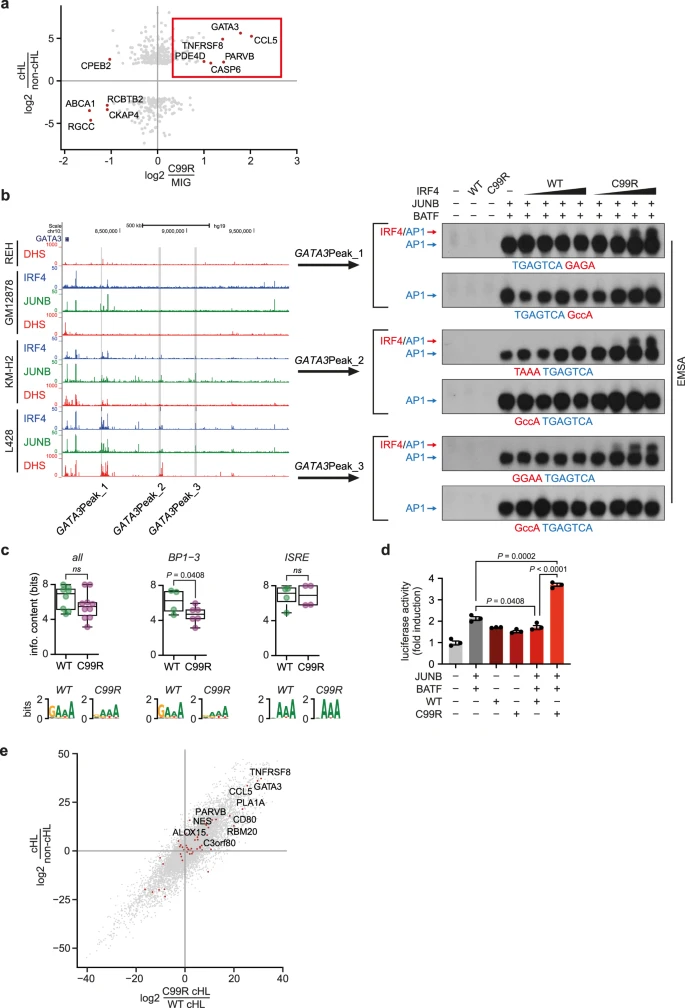
圖4:IRF4-C99R以非經典AICE2依賴的方式上調包含cHL標志性的基因
結論
在這里,作者描述了人類淋巴瘤,特別是cHL中TF改變的獨特機制,涉及靶向IRF4的DNA結合域中心的復發性體細胞錯義突變c.295 T>c(p.Cys99Arg;p.C99R)。他們發現,IRF4-C99R導致IRF4的DNA結合特性發生根本性變化,結合到經典IRF基序的結合損失和結合到經典和非經典IRF CE的新形態獲得。在功能上,證明IRF4-C99R阻斷了IRF4依賴性漿細胞誘導,并以非經典激活蛋白-1(AP-1)-IRF-CE(AICE)依賴的方式上調疾病特異性基因。
實驗方法
細胞培養,細胞轉染,電泳遷移率變化分析(EMSA),免疫印跡,桑格測序,PCR,免疫組化,重組蛋白的克隆與純化,ChIP-seq,DNase-seq,RNA-seq,流式細胞術
參考文獻
Schleussner N, Cauchy P, Franke V, Giefing M, Fornes O, Vankadari N, Assi SA, Costanza M, Weniger MA, Akalin A, Anagnostopoulos I, Bukur T, Casarotto MG, Damm F, Daumke O, Edginton-White B, Gebhardt JCM, Grau M, Grunwald S, Hansmann ML, Hartmann S, Huber L, K?rgel E, Lusatis S, Noerenberg D, Obier N, Pannicke U, Fischer A, Reisser A, Rosenwald A, Schwarz K, Sundararaj S, Weilemann A, Winkler W, Xu W, Lenz G, Rajewsky K, Wasserman WW, Cockerill PN, Scheidereit C, Siebert R, Küppers R, Grosschedl R, Janz M, Bonifer C, Mathas S. Transcriptional reprogramming by mutated IRF4 in lymphoma. Nat Commun. 2023 Nov 7;14(1):6947. doi: 10.1038/s41467-023-41954-8. PMID: 37935654; PMCID: PMC10630337.