






SUCLG2通過琥珀酰化調控肺腺癌線粒體功能障礙
線粒體功能障礙和能量代謝異常是癌癥的主要特征。然而,癌癥進展過程中線粒體功能障礙的機制遠未闡明。本文證明,琥珀酰輔酶A(CoA)合成酶GDP形成亞基β(SUCLG2)的表達水平會影響肺腺癌(LUAD)細胞的整體琥珀酰化。琥珀酰化組分析表明,缺失SUCLG2可上調線粒體蛋白的琥珀酰化水平,并通過降低酶活性或蛋白穩定性來抑制關鍵代謝酶的功能,從而抑制LUAD細胞的線粒體功能。有趣的是,SUCLG2本身也會在Lys93上發生琥珀酰化,這種琥珀酰化會增強其蛋白質的穩定性,從而導致SUCLG2的上調,促進LUAD細胞的增殖和腫瘤發生。Sirtuin 5(SIRT5)對SUCLG2的Lys93進行脫琥珀酰化,然后由含三方基序蛋白21(TRIM21)通過K63連接介導泛素化,并在溶酶體中降解。研究結果揭示了SUCLG2在線粒體功能障礙中的新作用,闡明了SUCLG2在LUAD中琥珀酰化介導的蛋白質平衡機制,從而為開發針對SUCLG2的抗癌藥物提供了理論依據。本文于2023年10月發表于《Advanced Science》,IF=15.1。
技術路線
實驗結果
1. SUCLG2是LUAD細胞增殖的必要條件,與LUAD患者的不良生存率密切相關
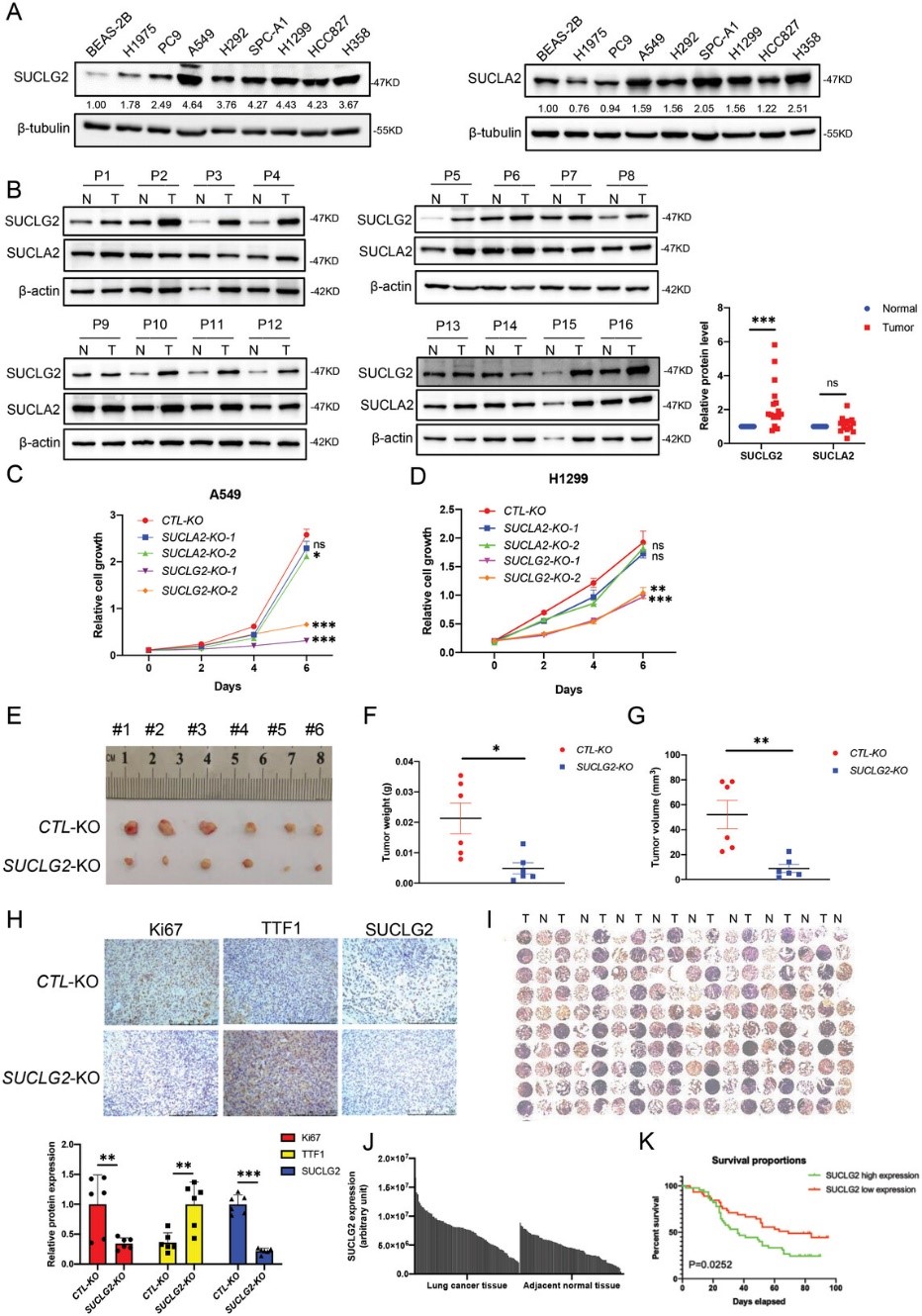
GDP特異性琥珀酰-CoA合成酶(SUCLG2)和ATP特異性琥珀酰-CoA合成酶(SUCLA2)是琥珀酰-CoA合成酶的兩個不同的β亞基。為了研究SUCL在LUAD 中的作用,作者首先檢測了SUCLG2和SUCLA2在LUAD細胞和組織中的蛋白表達,作者發現,與人類支氣管上皮細胞系BEAS-2B相比,SUCLG2在所有LUAD細胞中的蛋白表達均顯著上調。與BEAS-2B相比,SUCLA2在一些LUAD細胞中上調,但在另一些細胞中則下調,而且未發現一致的趨勢(圖1A)。當作者檢測LUAD組織中SUCLG2和SUCLA2的表達時,也得到了類似的結果。圖1B顯示,SUCLG2在LUAD組織中的表達高于鄰近的正常組織,而SUCLA2在大多數組織對中無明顯變化。為了研究SUCLG2和SUCLA2在LUAD細胞增殖中的功能,作者使用CRISPR-Cas9技術刪除了A549和H1299細胞中的SUCLG2或SUCLA2。然后,作者進行了細胞增殖實驗,當SUCLG2而非SUCLA2被敲除時,LUAD細胞的增殖率明顯下降(圖1C,D)。因此,這些結果表明,SUCLG2 而不是SUCLA2在LUAD中起著重要作用。
作者還進行了異種移植試驗,以確定A549野生型(WT)和SUCLG2基因敲除(SUCLG2-KO)細胞的腫瘤發生情況。與A549-WT細胞相比,A549 SUCLG2-KO的腫瘤重量和體積均有所下降(圖1E-G)。為了證實這些結果,作者使用抗Ki67、抗TTF1和抗SUCLG2抗體對A549-WT和A549-SUCLG2-KO的腫瘤樣本進行了免疫組化(IHC)分析。Ki67在病理評估中被廣泛用作增殖標志物,而TTF1在高級別LUAD中經常被抑制。作者的結果顯示,A549-WT組的Ki67和SUCLG2染色明顯高于A549-SUCLG2-KO組。然而,A549-WT組的TTF1染色低于A549-SUCLG2-KO組(圖1H)。為了進一步確定SUCLG2在LUAD中的高表達是否具有臨床意義,作者使用抗SUCLG2抗體通過IHC檢測了SUCLG2在LUAD組織陣列中的蛋白表達。該組織陣列包括來自90名LUAD患者的肺癌組織和鄰近的正常組織。結果表明,與鄰近的正常組織相比,腫瘤組織表達的SUCLG2水平明顯更高(圖1I);染色定量進一步驗證了這一結果(圖1J)。對癌癥組織染色定量結果的統計分析根據SUCLG2水平將樣本分為兩組。患者生存率與 SUCLG2 水平相關。如圖1K所示,SUCLG2水平低的患者比SUCLG2水平高的患者生存率更高(P = 0.0252)。這些結果表明,SUCLG2影響LUAD細胞的增殖和腫瘤發生,并與LUAD的進展和患者生存密切相關。
2. SUCLG2 缺乏導致線粒體功能障礙
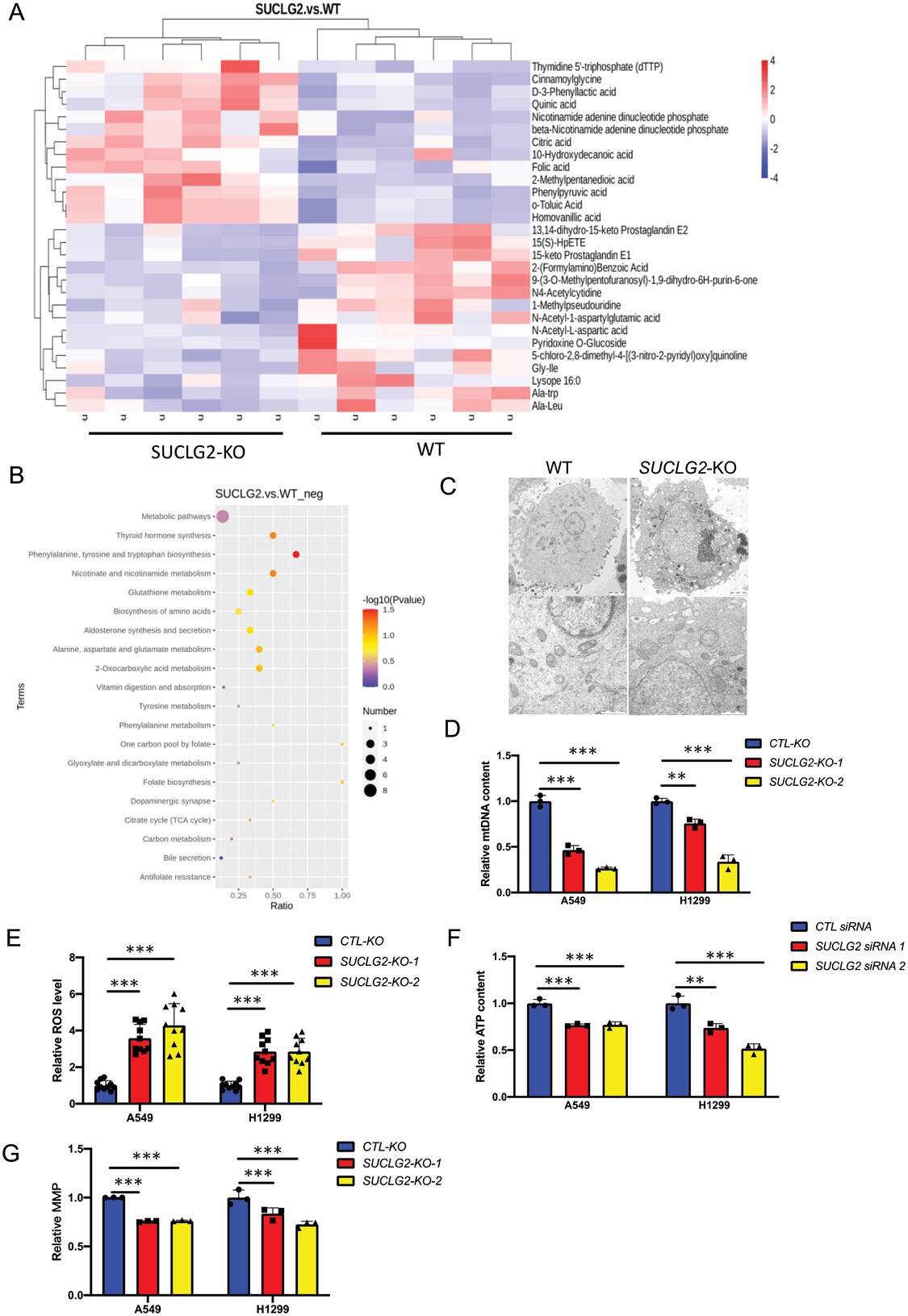
線粒體是新陳代謝活動的核心細胞器,而SUCLG2是TCA循環中的一個關鍵酶。因此,作者推測SUCLG2可能會通過調節線粒體代謝來影響腫瘤的增殖。作者對A549-WT和A549-SUCLG2-KO細胞進行了非靶向代謝組學研究,發現與A549-WT細胞相比,A549-SUCLG2-KO細胞中有15種代謝物上調,64種代謝物下調(圖2A)。作者利用KEGG通路分析對代謝物進行了分析,發現線粒體相關代謝通路如TCA循環和谷胱甘肽代謝發生了顯著變化(圖2B)。為了檢測SUCLG2對線粒體功能的影響,作者進行了透射電子顯微鏡(TEM)檢查,結果發現,A549細胞中的線粒體呈管狀,嵴清晰,而A549-SUCLG2-KO細胞中的線粒體腫脹,嵴斷裂(圖2C)。作者發現在A549和H1299細胞中敲除SUCLG2會降低mtDNA水平(圖2D)。此外,與對照細胞相比,SUCLG2-KO 細胞中的細胞ROS水平升高(圖2E)。敲除SUCLG2后,A549和H1299細胞中的ATP含量下降(圖2F)。隨后,作者測量了敲除SUCLG2 的A549和H1299細胞的線粒體膜電位(MMP)。與對照細胞相比,SUCLG2基因敲除細胞的線粒體膜電位明顯降低(圖2G)。從這些結果中,作者得出結論,LUAD細胞線粒體功能的維持依賴于SUCLG2的表達。
3. SUCLG2基因敲除誘導蛋白質琥珀酰化的全面增加
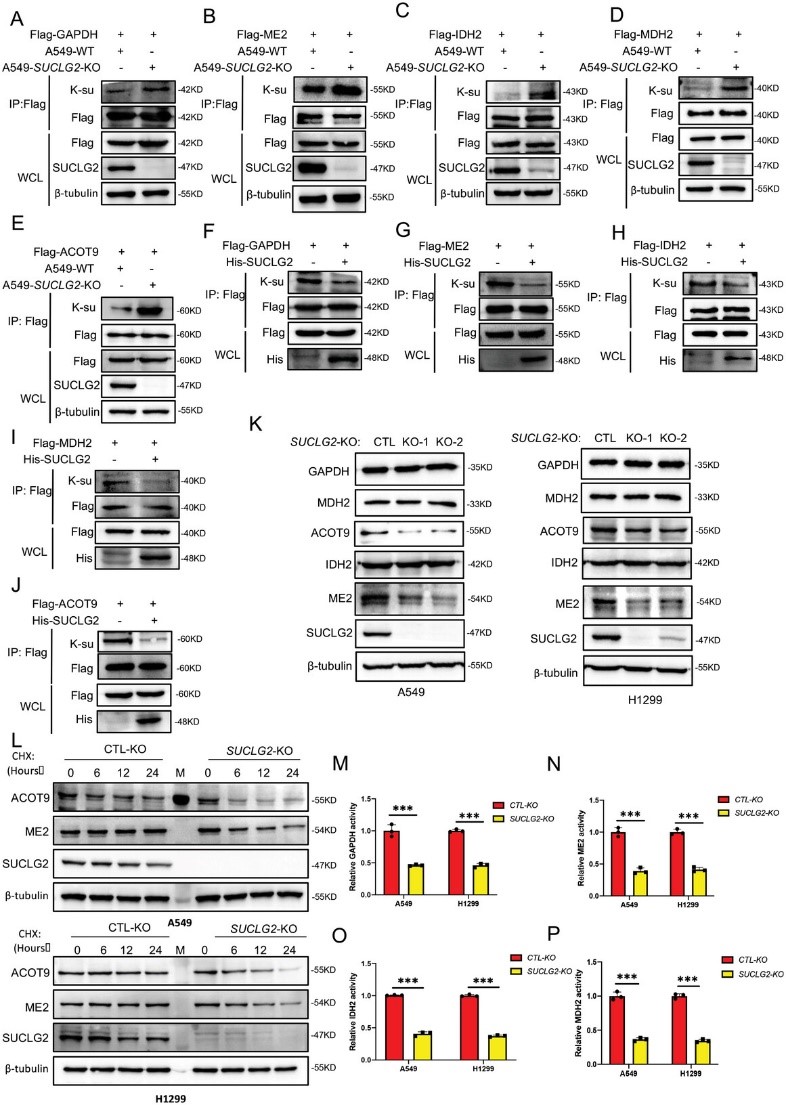
SUCLG2是琥珀酰-CoA的主要水解酶,而琥珀酰-CoA是蛋白質琥珀酰化的主要琥珀酰修飾底物。琥珀酰化被認為是一種受琥珀酰-CoA供體濃度調節的非酶促反應。因此,作者推測SUCLG2可能通過琥珀酰化調節線粒體功能和代謝重編程。由于琥珀酰-CoA會被氧化水解,因此很難檢測到。因此,作者在敲除SUCLG2的A549和 H1299細胞中檢測了由SUCLG2催化的琥珀酰-CoA產物琥珀酸的含量。圖3A顯示,SUCLG2基因敲除明顯降低了琥珀酸的含量。接下來作者研究了SUCLG2對細胞內蛋白質琥珀酰化水平的影響。結果顯示,SUCLG2基因敲除增加了細胞內蛋白質的琥珀酰化水平(圖3B)。然后,作者進行了琥珀酰4D質譜分析,以進一步研究受SUCLG2調控的蛋白質的琥珀酰化情況。結果發現,在SUCLG2基因敲除的細胞中,285個蛋白質中有686個琥珀酰賴氨酸(K-su)位點,44個蛋白質中有61個不同表達的琥珀酰化位點。作者發現,與A549-WT細胞相比,在A549-SUCLG2-KO細胞中發現的大部分琥珀酰化上調,61%分布在線粒體中(圖3C)。GO分析表明,線粒體相關的代謝途徑,如能量產生和轉化,發生了顯著變化(圖3D)。與線粒體功能相關的甘油醛-3-磷酸脫氫酶(GAPDH)、NAD依賴性蘋果酸酶(ME2)、異檸檬酸脫氫酶(IDH2)、蘋果酸脫氫酶(MDH2)和酰輔酶 A 硫代酯酶9(ACOT9)等關鍵酶的琥珀酰化在特定位點顯著上調(圖3E-J)。這些結果表明,SUCLG2基因敲除促進了與線粒體功能相關的蛋白質的琥珀酰化。

4. SUCLG2通過調節琥珀酰化影響蛋白質的穩定性或酶活性
接下來,作者研究了SUCLG2對GAPDH、ME2、IDH2、MDH2和ACOT9等關鍵酶功能的影響。作者首先檢測了SUCLG2對這些蛋白質琥珀酰化的影響,以驗證琥珀酰4D質譜分析結果。結果顯示,SUCLG2敲除會增加GAPDH、ME2、IDH2、MDH2和ACOT9的琥珀酰化(圖4A-E),而在A549細胞中過表達SUCLG2會降低這些蛋白的琥珀酰化(圖4F-J)。這些結果證實,SUCLG2調控線粒體相關蛋白的琥珀酰化。接下來,作者研究了SUCLG2如何影響這些代謝酶的功能。首先,作者用Western印跡法檢測了SUCLG2敲除的A549和H1299細胞中GAPDH、ME2、IDH2、MDH2和ACOT9的表達。圖4K顯示,SUCLG2基因敲除降低了ME2和ACOT9的蛋白表達,而GAPDH、IDH2和MDH2的表達未受影響。為了確定SUCLG2如何調控ME2和ACOT9的表達,作者測定了A549-WT和A549-SUCLG2-KO細胞中ME2和ACOT9的穩定性。結果顯示,與A549-WT細胞相比,ME2和ACOT9蛋白在A549-SUCLG2-KO細胞中的降解速度更快(圖4L)。為了進一步證實蛋白質的穩定性受琥珀酰化的調控,作者對ACOT9(K157R,K294R)和ME2(K26R,K94R)的琥珀酰化位點進行了點突變,這些突變位點是通過琥珀酰4D質譜分析結果確定的。由于GAPDH、IDH2和MDH2的表達不受SUCLG2敲除的影響,作者接下來檢測了這些酶的酶活性。敲除SUCLG2會降低A549和H1299細胞中GAPDH、ME2、IDH2 和MDH2的活性(圖4M-P)。這些結果表明,敲除SUCLG2會降低酶活性或蛋白質穩定性,從而抑制與線粒體功能相關的關鍵代謝酶的功能。
5. TRIM21通過K63-連接泛素化介導的溶酶體途徑誘導SUCLG2降解
作者進行了環己亞胺(CHX)酶切實驗,檢測SUCLG2蛋白在LUAD細胞系(A549、HCC827和H1299)和BEAS-2B細胞中的穩定性。結果顯示,與A549、HCC827和H1299細胞相比,SUCLG2蛋白在BEAS-2B細胞中的降解速度明顯加快(圖5A)。這些結果表明,SUCLG2在LUAD中的高表達是由于蛋白質穩定性的增強。因此,作者研究了SUCLG2的降解途徑。作者發現,用CHX處理A549細胞24小時后,SUCLG2的蛋白水平明顯下降,加入溶酶體抑制劑氯喹(CQ)后可以恢復,但蛋白酶體抑制劑MG132不能恢復(圖5B)。此外,作者通過加入MG132或CQ檢測了SUCLG2的泛素化水平,發現加入CQ而不是MG132 時,SUCLG2的泛素化顯著增加(圖5C)。然后作者檢測了泛素化類型,發現在用CQ處理的A549細胞中,SUCLG2的K63-連接泛素化上調(圖5D)。這些結果表明,SUCLG2蛋白是通過K63鏈接泛素化介導的溶酶體途徑降解的。
為了確定SUCLG2的E3連接酶,作者進行了質譜分析,發現TRIM21是一種推定的SUCLG2相互作用蛋白(圖5E)。作者利用免疫沉淀技術驗證了 SUCLG2 與 TRIM21在A549細胞中的相互作用(圖5F)。然后,作者檢測了過表達或敲除TRIM21時SUCLG2 的蛋白水平。結果顯示,過表達TRIM21會降低A549細胞中SUCLG2蛋白的表達(圖5G)。此外,過表達TRIM21會加快CHX處理后SUCLG2的降解速度(圖5H)。同時,作者發現在LUAD細胞和BEAS-2B細胞中,TRIM21和SUCLG2的蛋白表達呈負相關(圖5I)。在LUAD組織中也觀察到了類似的趨勢。TRIM21的蛋白表達與SUCLG2、SIRT5和SUCLG2呈負相關,未見一致趨勢(圖5J)。作者進一步探討了TRIM21介導的SUCLG2泛素鏈連接類型,過表達TRIM21增加了 SUCLG2的K63連接泛素化(圖5K)。總之,這些數據支持 TRIM21 通過 K63 連接泛素化 SUCLG2,然后通過溶酶體降解的模型。
作者接下來確定了受TRIM21調控的SUCLG2泛素化位點。利用 PhosphoSitePlus,作者確定了K101、K118、K132、K200和K386為SUCLG2的推定泛素化位點(圖 5L)。然后,作者構建了含有指定突變(K101R、K118R、K132R、K200R和K386R)的SUCLG2質粒。作者發現,當TRIM21被過表達時,只有SUCLG2K200R的泛素化沒有發生變化(圖5M)。接下來,作者測量了野生型SUCLG2(SUCLG2WT)和 SUCLG2K200R 突變體的穩定性。在用CHX處理細胞時,SUCLG2K200R 的降解速度比 SUCLG2WT 慢得多。此外,TRIM21的過表達并不能提高SUCLG2K200R的降解率(圖5N)。這些數據表明,SUCLG2的K200是TRIM21的主要泛素化位點。
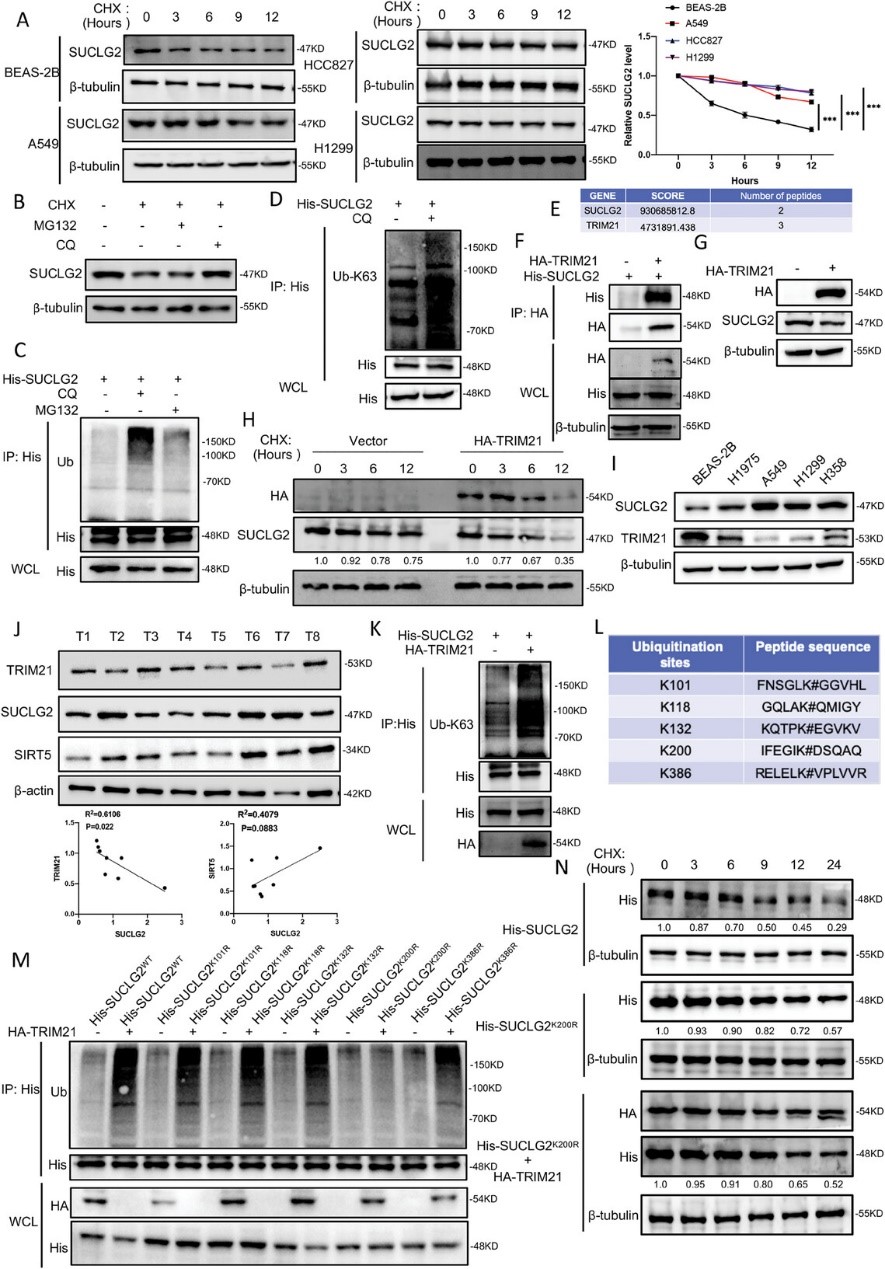
6. SUCLG2的K93-琥珀酰化影響TRIM21介導的SUCLG2降解
SUCLG2定位于線粒體基質,是將琥珀酰-CoA轉化為琥珀酸的關鍵。因此,作者推測琥珀酰化會影響SUCLG2的功能。首先,作者利用共轉印證明了SUCLG2蛋白可被琥珀酰化(圖6A)。利用PhosphoSitePlus和質譜分析結果,作者確定K78、K93和K338為SUCLG2的假定琥珀酰化位點(圖6B)。為了確定SUCLG2的主要琥珀酰化位點,作者構建了含有所述突變(K78R、K93R 和 K338R)的SUCLG2質粒,發現與SUCLG2WT相比,只有SUCLG2K93R的琥珀酰化減少(圖6C)。接下來作者測量了SUCLG2WT和SUCLG2K93R的穩定性。用CHX處理細胞時,SUCLG2K93R蛋白水平的下降速度比SUCLG2WT快(圖6D)。這些結果表明,SUCLG2的K93是SUCLG2的主要琥珀酰化位點,其突變會降低蛋白在LUAD細胞中的穩定性。
作者接下來探討了SUCLG2的琥珀酰化是否會調控其泛素化。作者進行了聯合泛素化檢測,發現與SUCLG2WT相比,SUCLG2K93R的泛素化作用更強(圖6E)。這一結果表明,SUCLG2K93R突變體影響了SUCLG2的泛素化。作者還發現TRIM21更傾向于結合SUCLG2K93R并增加其泛素化(圖6F和G)。此外,與SUCLG2WT相比,SUCLG2K93R結合了更多的p62(圖 6H)。這些結果證實,SUCLG2K93R通過增加 TRIM21介導的泛素化和溶酶體降解來促進其降解。
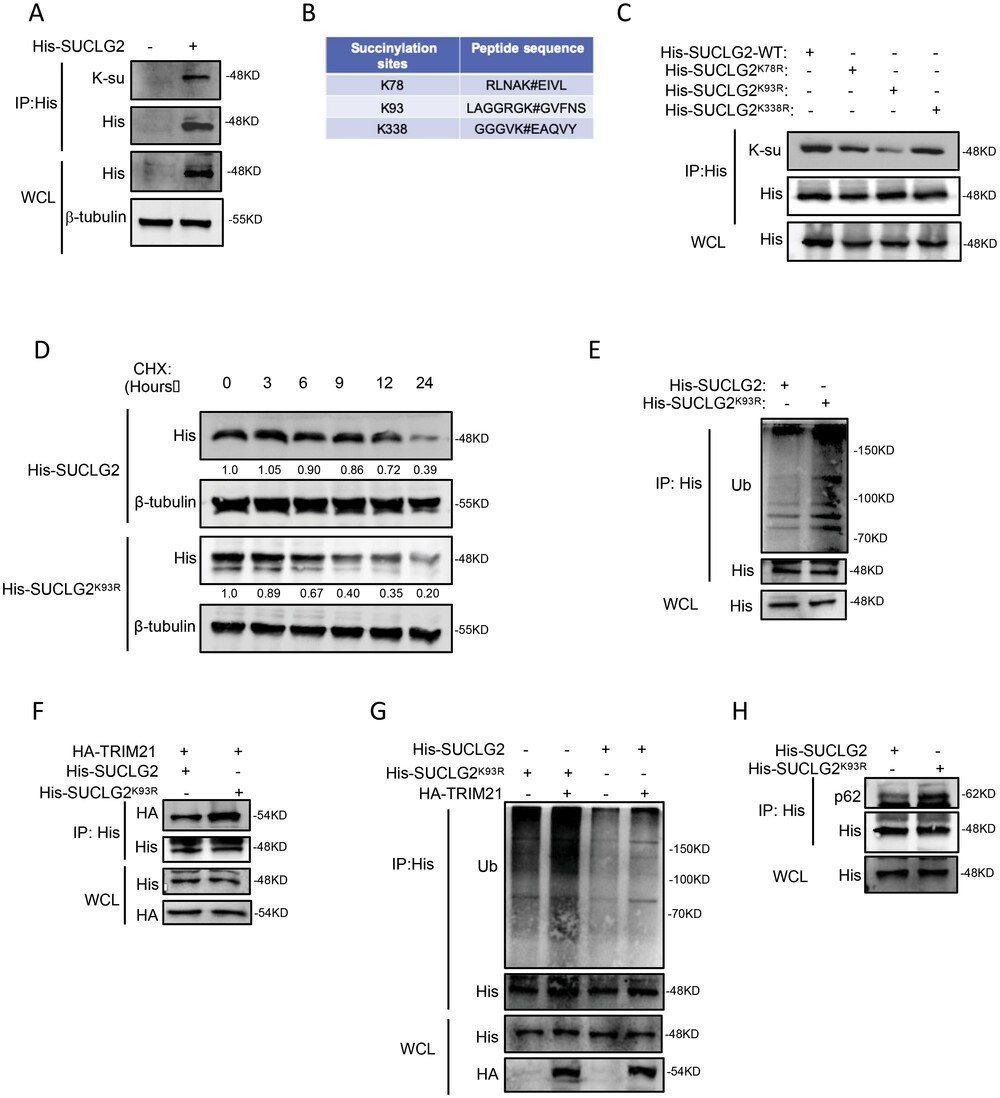
7. SIRT5 是SUCLG2的脫琥珀酰化酶并影響其蛋白穩定性
脫琥珀酰化主要由依賴NAD+ 的SIRT家族調控。作者試圖確定哪種SIRT參與了SUCLG2的脫琥珀酰化。迄今為止,已有報道稱SIRT5和SIRT7是線粒體中的脫琥珀酰化酶。作者研究了SIRT5和SIRT7是否能使SUCLG2去琥珀酰化并影響其功能。在過表達SIRT5的細胞中,琥珀酰化的SUCLG2水平明顯低于含有空載體的細胞(圖7A)。圖7B顯示,敲除SIRT5增加了SUCLG2的琥珀酸化。接下來,作者通過共顯微鏡(co-IP)驗證了SUCLG2和SIRT5之間的相互作用。結果顯示,SUCLG2 和SIRT5相互作用(圖 7C、D)。為了進一步驗證它們之間的相互作用,作者進行了免疫熒光和線粒體分離試驗。結果顯示,SIRT5與SUCLG2在線粒體中共位(圖 7E和F),表明SIRT5與SUCLG2相互作用。
作者接下來探討了SIRT5和SUCLG2之間的關系。首先,過表達或敲除SIRT5,并檢測SUCLG2的表達。敲除SIRT5導致SUCLG2蛋白水平升高,而過表達SIRT5 則降低了SUCLG2的表達(圖7G、H)。利用CHX阻斷SUCLG2的蛋白合成,作者發現過表達SIRT5會加速SUCLG2的降解(圖7I)。接著,作者發現過表達SIRT5 會顯著增加SUCLG2的泛素化水平(圖7J),而敲除SIRT5則會減少SUCLG2的泛素化(圖7K)。作者進一步檢測了SIRT5誘導的SUCLG2泛素鏈的類型。過表達 SIRT5增加了SUCLG2的K63連接泛素化(圖7L)。作者還利用共轉錄分析發現,在A549細胞中,過表達SIRT5可促進SUCLG2與TRIM21和p62的結合(圖7M,N)。這些結果表明,SIRT5通過K63連接泛素化SUCLG2,然后通過溶酶體途徑降解。
接下來,作者探討了SIRT5與SUCLG2K93R 之間的關系。作者發現SIRT5并不影響SUCLG2K93R的琥珀酰化(圖7O)。綜上所述,這些結果表明SIRT5是SUCLG2 在Lys93上的脫琥珀酰化酶,促進了TRIM21介導的SUCLG2降解。
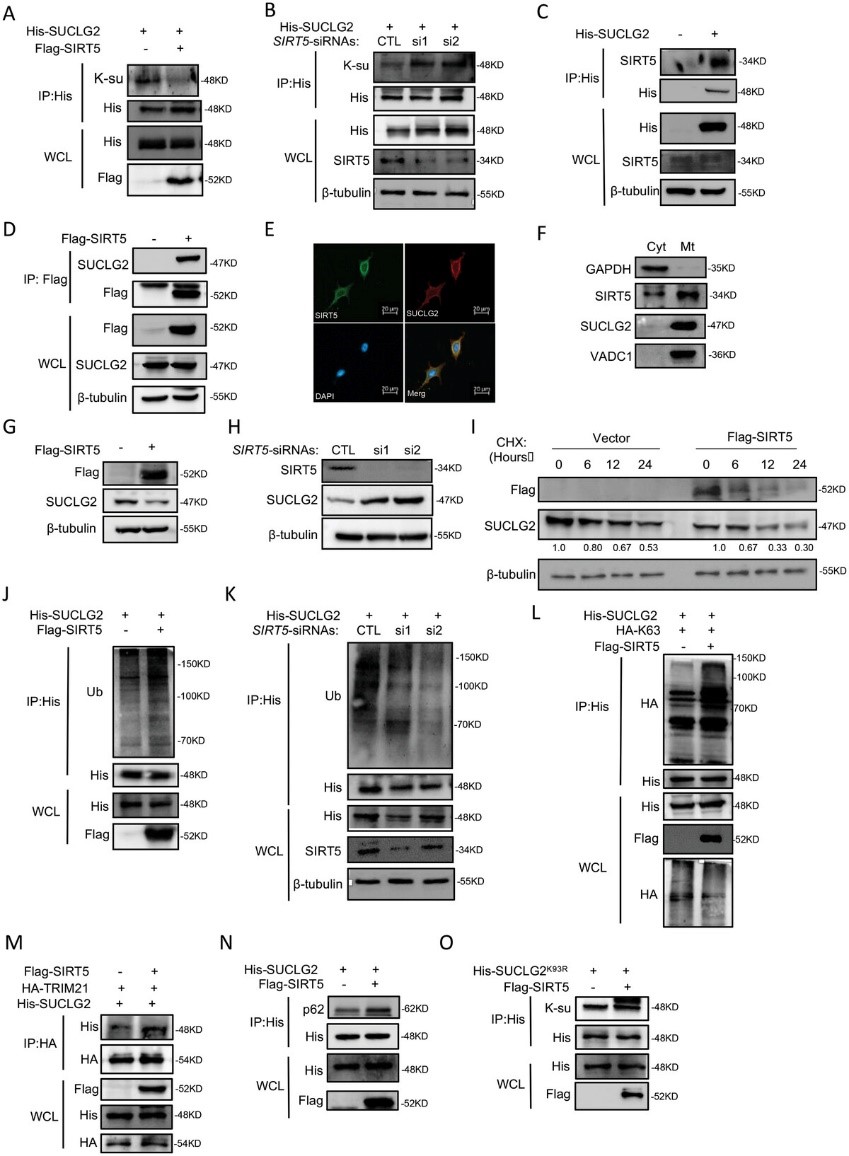
8. 抑制SUCLG2在K93上的琥珀酰化會導致LUAD細胞線粒體功能障礙并減少腫瘤發生
為了進一步研究TRIM21是否通過調控SUCLG2的泛素化和降解來影響腫瘤的發生,作者在A549和H1299細胞中過表達了TRIM21和SUCLG2,細胞生長實驗顯示,過表達TRIM21抑制了A549和H1299細胞的生長。然而,SUCLG2的表達克服了過表達TRIM21對細胞增殖的抑制作用(圖8A,B)。同樣,SIRT5也會抑制A549和H1299細胞的生長,而SUCLG2會降低過表達SIRT5對細胞生長的抑制作用(圖8C和D)。這些結果表明,SUCLG2介導了SIRT5和TRIM21對LUAD細胞增殖的功能。為了給開發靶向SUCLG2的抗腫瘤藥物提供理論依據,作者去除了內源性SUCLG2,并將野生型SUCLG2(SUCLG2WT)或SUCLG2K93R突變體穩定地重新導入A549細胞(圖8E)。作者評估了SUCLG2K93R 突變體對線粒體功能的影響,并使用 TEM觀察線粒體的形態變化。A549-SUCLG2WT細胞中的線粒體嵴完整,形態特征正常。然而,A549-SUCLG2K93R細胞中的線粒體出現空泡化和腫脹,嵴斷裂(圖8F)。為了進一步確定線粒體質量的差異,作者檢測了A549-SUCLG2WT和A549-SUCLG2K93R細胞的mtDNA拷貝數。作者發現,與A549-SUCLG2WT細胞相比,A549-SUCLG2K93R細胞的mtDNA拷貝數減少了(圖8G)。作者還發現,與 A549-SUCLG2WT 細胞相比,ROS 在 A549-SUCLG2K93R 細胞中明顯積累(圖 8H)。A549-SUCLG2WT細胞中的ATP含量高于A549-SUCLG2K93R細胞(圖8I)。作者的研究結果表明,A549-SUCLG2WT 的線粒體功能優于 A549-SUCLG2K93R。接下來,作者在 A549-SUCLG2WT 和 A549-SUCLG2K93R 細胞中進行了細胞增殖試驗。結果顯示,SUCLG2K93R 突變體抑制了細胞增殖(圖 8J)。然后,作者在異種移植模型中評估了 A549-SUCLG2K93R 細胞的腫瘤發生情況。與A549-SUCLG2WT相比,A549-SUCLG2細胞的異種移植物顯示腫瘤重量和體積減少(圖8K-M),IHC結果顯示,與SUCLG2WT腫瘤相比,SUCLG2K93R突變體腫瘤的Ki67表達減少,TTF1表達增加(圖8N)。這些結果表明,抑制SUCLG2在K93上的琥珀酰化會導致線粒體功能障礙,減少LUAD細胞的腫瘤發生。
結論:
綜上所述,作者的研究闡明了SUCLG2在線粒體功能障礙中的作用,闡明了SUCLG2在LUAD中琥珀酰化介導的蛋白平衡機制,從而為開發靶向SUCLG2的抗腫瘤藥物提供了理論依據。
實驗方法:
RT-qPCR檢測、免疫印跡和蛋白質印跡、免疫熒光檢測、免疫組化、mtDNA提取和分析、ATP生產測量、線粒體和胞質溶膠提取、MDH2活性的測量、IDH2活性的測量、ME2活性的測量、GAPDH活性的測量、琥珀酰基4D質譜分析、非靶向代謝組學、細胞培養和轉染、細胞增殖檢測、transwell遷移檢測和劃痕傷口愈合檢測、CRISPR-Cas9介導的基因破壞、細胞內ROS檢測、體內異種移植檢測
參考文獻:
Hu, Q., Xu, J., Wang, L., Yuan, Y., Luo, R., Gan, M., Wang, K., Zhao, T., Wang, Y., Han, T., Wang, J.-B., SUCLG2 Regulates Mitochondrial Dysfunction through Succinylation in Lung Adenocarcinoma. Adv. Sci. 2023, 2303535.