






靶向PGLYRP1促進抗腫瘤免疫同時抑制自身免疫性神經炎癥
摘要
共抑制和檢查點分子抑制腫瘤微環境中T細胞的功能,從而使T細胞失去功能。盡管免疫檢查點阻滯是多種人類癌癥的成功治療選擇,但嚴重的自身免疫樣不良效應可能會限制其應用。在這里,我們展示了編碼肽聚糖識別蛋白1(PGLYRP1)的基因高度與編碼共抑制分子的基因共表達,這表明它可能是癌癥免疫治療的有前途的靶點。在小鼠中遺傳敲除Pglyrp1導致腫瘤生長減緩,并使CD8+ T細胞呈現出增強的激活/效應器表型,這表明PGLYRP1在CD8+ T細胞中具有抑制功能。令人驚訝的是,遺傳敲除Pglyrp1能夠保護免受實驗性自身免疫腦脊髓炎的發展,這是中樞神經系統自身免疫疾病的模型。PGLYRP1缺陷的髓系細胞在抗原呈遞和T細胞激活方面存在缺陷,這表明PGLYRP1在自身免疫期間可能在髓系細胞中起到促炎分子的作用。這些結果突出了PGLYRP1作為免疫治療的有前途的靶點,當被靶向時,能夠引發強大的抗腫瘤免疫應答,同時保護免受一些形式的組織炎癥和自身免疫疾病的影響。
該研究于2023年10月發表在《Nature Immunology》,IF:30.5。
技術路線

結果
1、Plyrp1與共抑制基因在T細胞中的共表達
我們實驗室以及其他研究組的早期工作表明,在T細胞中,共抑制分子高度共表達在一個基因模塊中。因此,我們推斷,與共抑制基因在腫瘤中CD8+ T細胞中高度共表達的基因可能編碼新的T細胞檢查點分子。我們利用了我們先前發表的CD8+腫瘤浸潤淋巴細胞(TILs)的單細胞RNA測序(scRNA-seq)數據,這些數據來自B16F10黑色素瘤,以識別共變化的所有基因通過單個細胞的編碼已知免疫檢查點(Pdcd1、Tigit、Ctla4、Havcr2和Lag3)的基因與干細胞特性相關的基因(Ccr7、Cxcr5、Tcf7和Sell)(圖1a)。T細胞中編碼多種已知功能分子的基因,包括Cxcr6、Nkg7、Gzmb、Ifng、Prf1、Irf8和Bhlhe40,與檢查點基因正相關并與干細胞基因負相關。此外,Pglyrp1一個在T細胞中沒有已知功能的基因,也遵循這樣的模式(與共抑制分子的Spearman相關系數=0.47,P=1.2×10^-21;與干細胞基因的Spearman相關系數= -0.27,P=1.8×10^-7;P值是用Spearman的漸近t檢驗計算的)。一致地,Pglyrp1在表達共抑制基因(Pdcd1、Tigit、Ctla4、Havcr2和Lag3)和T細胞功能障礙特征的細胞群中表達,而在表達干細胞樣基因(Slamf6、Ccr7、Cxcr5、Tcf7和Sell)的細胞群中不表達(圖1b、c)。我們通過實驗證實,從MC38-OVA腫瘤中分離的PD-1+TIM-3+ CD8+ T細胞,這些細胞構成了耗竭T細胞,Pglyrp1的表達在體外增加(圖1d)。在對來自人類黑色素瘤的免疫細胞進行單細胞RNA測序分析時,我們發現PGLYRP1與其他共抑制分子一起在耗竭CD8+ T細胞中表達最高,盡管其RNA水平較低于編碼其他檢查點分子的RNA,這表明PGLYRP1與共抑制基因的共表達在耗竭人類T細胞中也是保守的,但可能由于RNA水平較低而沒有得到足夠的關注。
因此,我們的數據顯示,Pglyrp1在小鼠和人類的耗竭CD8+ T細胞中與共抑制基因高度共表達。

2、IL-27對T細胞Plyrp1表達的調節
我們先前已經證明,細胞因子白細胞介素-27(IL-27)誘導了T細胞中的一個共抑制模塊。值得注意的是,Pglyrp1是這個由IL-27誘導的共抑制模塊的一部分。為了確認IL-27在T細胞中誘導Pglyrp1表達,我們體外培養了有或沒有IL-27原生的CD4+和CD8+ T細胞,確認IL-27信號傳導后CD4+(約27倍)和CD8+(約2.5倍)T細胞中Pglyrp1表達顯著增加(圖1e)。先前已經確定轉錄因子PRDM1和c-MAF是IL-27誘導的共抑制模塊的協同調控因子。為了測試它們是否對IL-27介導的T細胞中Pglyrp1表達的誘導起作用,我們體外培養了來自PRDM1/c-MAF缺陷小鼠(Maf–/–Prdm1–/–)的原生的有或沒有IL-27的CD4+ T細胞(圖1f)。結果顯示,IL-27對Pglyrp1表達的誘導在Maf–/– Prdm1–/– T細胞中明顯減少,表明IL-27-PRDM1/c-MAF軸在體外誘導T細胞中的Pglyrp1表達。為了檢查IL-27和PRDM1/c-MAF是否在T細胞中的Pglyrp1表達中起作用,我們分析了先前發表的CD8+ T細胞的總RNA測序數據,這些細胞是從野生型(WT)和IL-27受體缺陷型(Il27ra–/–)小鼠或Maf–/–Prdm1–/–小鼠的B16F10黑色素瘤腫瘤中分離的(圖1g,h)。在這兩種情況下,與相應的WT對照相比,我們發現Pglyrp1的表達水平降低。因此,我們的結果表明,IL-27-PRDM1/c-MAF軸在體內外都調控T細胞中Pglyrp1的表達。
3、Plyrp1基因缺陷小鼠中腫瘤生長受到抑制
為了分析PGLYRP1在抗腫瘤免疫中是否起到功能性作用,我們研究了PGLYRP1在不同人類癌癥中的表達水平與生存率之間的關聯,并發現高表達PGLYRP1的腫瘤患者有明顯更差的預后,這表明PGLYRP1在人類癌癥中可能作為抗腫瘤免疫的負調控因子。
為了研究PGLYRP1在免疫反應中的作用,我們獲得了Pglyrp1–/–小鼠,這些小鼠在穩態下對免疫系統組成和T細胞表型沒有改變。為了測試PGLYRP1在小鼠抗腫瘤免疫中的作用,我們植入MC38-OVA結腸癌細胞和B16-OVA黑色素瘤細胞到Pglyrp1–/–小鼠和相匹配的野生型(WT)同窩對照小鼠中(圖2)。我們發現植入MC38-OVA細胞的Pglyrp1–/–小鼠的腫瘤大小和腫瘤重量減小(圖2a–c),這表明PGLYRP1可能是抗腫瘤免疫的潛在負調控因子。我們還發現Pglyrp1–/–小鼠植入B16-OVA黑色素瘤細胞的腫瘤生長減緩,突顯了PGLYRP1缺陷對不同腫瘤類型的腫瘤生長具有保守性作用。
為了了解PGLYRP1可能在腫瘤微環境中對哪種免疫細胞類型發揮作用,我們通過實時定量PCR(qPCR)評估了MC38-OVA腫瘤中分離的不同免疫細胞群體中Pglyrp1的表達。與我們先前的分析一致,Pglyrp1的表達在耗竭CD8+ T細胞中最高。在腫瘤微環境的固有成分中,自然殺傷細胞(NK細胞)和中性粒細胞表達Pglyrp1的水平最高。在MC38-OVA腫瘤中,相對頻率方面,Pglyrp1–/–小鼠與野生型小鼠相比,免疫細胞種群沒有主要差異,除了Treg細胞在Pglyrp1–/–小鼠的腫瘤中明顯減少(圖2d)。
因為Pglyrp1與CD8+ T細胞中的共抑制分子共表達,我們接下來集中研究CD8+ T細胞組分。有趣的是,Pglyrp1–/–小鼠的腫瘤和淋巴結中CD8+ T細胞中高頻率表達促炎細胞因子,包括干擾素-γ(IFNγ)、腫瘤壞死因子(TNF)和粒細胞-巨噬細胞集落刺激因子(GM-CSF),表明增加了效應功能(圖2e)。我們在CD4+ T細胞中沒有發現細胞因子表達的差異,除了腫瘤中IFNγ產生的增加。TIM-3和PD-1是共抑制分子,在T細胞激活過程中上調,也是終末耗竭T細胞的標志。我們發現Pglyrp1–/–小鼠的腫瘤、淋巴結和脾臟中CD8+ T細胞中TIM-3和PD-1的表達增加,TIM-3+ PD-1+ CD8+ T細胞的頻率也增加(圖2f,g)。我們在CD4+ T細胞的TIM-3和PD-1表達沒有發現差異。結合我們的功能數據,這些發現表明Pglyrp1–/–小鼠的CD8+ TILs的激活和效應表型增強。
為了全面描述Pglyrp1–/–小鼠中CD8+ T細胞的表型,我們對Pglyrp1–/–小鼠和相匹配的WT同窩對照中的腫瘤浸潤CD8+ T細胞進行了bulk RNA測序。我們鑒定了在Pglyrp1–/– 與WT CD8+ T細胞中上調的330個基因和下調的35個基因(FDR<0.15和|log2(fold change)|>1;P值是用edgeR中的似然比檢驗計算的,經過Benjamini-Hochberg方法(FDR)進行了多重比較調整;圖2h)。與我們的流式分析一致,Pglyrp1–/–與WT CD8+ T細胞中表達增加的基因包括參與T細胞激活、效應功能和耗竭基因(Nr4a2、Prf1、Lag3、Irf8、Atf3、Ifng、Havcr2、Tigit和Maf;圖2h,i),并富集在T細胞效應和耗竭標志中(圖2j,k)。

4、Pglyrp1–/– CD8+ TILs 顯示向效應細胞/耗竭細胞轉變
為了更好地理解Pglyrp1–/– CD8+ TILs中共抑制分子和效應功能同時增加,我們對來自Pglyrp1–/–小鼠和相匹配的WT對照小鼠的CD45+ CD3+ TILs進行了scRNA測序(圖3)。細胞聚類成四個亞群(圖3a),我們根據標記物表達將其注釋為Treg細胞、CD4+常規T(Tconv)細胞和兩個CD8+ T細胞簇。在其中一個CD8+ T細胞簇中,包括與WNT和TCF信號傳導以及干細胞和原始樣細胞相關的通路富集,表示這個簇為干細胞樣細胞。相反,另一個CD8+ T細胞簇富集了T細胞激活、效應功能和耗竭標志,表明一個簇為干細胞樣細胞,另一個簇為效應器/耗竭細胞。RNA速度分析表明從干細胞樣細胞到效應器/耗竭細胞的軌跡,進一步支持這一模型。與我們早期的分析一致(圖1),Pglyrp1的表達在效應器/耗竭和Treg細胞簇中最高(圖3b)。
WT小鼠和Pglyrp1–/–小鼠的細胞特征在Treg細胞和CD4+ Tconv細胞簇中混合良好,只有少數差異表達的基因(在Treg細胞和CD4+ Tconv細胞中分別上調8和41個基因,下調4和7個基因;FDR<0.05和|log2(fold change)|≥0.25;P值是用edgeR中的經驗Bayes準似然F檢驗計算的,經過Benjamini-Hochberg方法(FDR)進行了多重比較調整;圖3c)。與我們的流式分析一致(圖2d),Pglyrp1–/–小鼠的腫瘤中Treg細胞的比例顯著減少(圖3d)。在WT小鼠和Pglyrp1–/–小鼠的Treg細胞之間差異表達的少數基因中,Itgae(編碼CD103)是Pglyrp1–/–小鼠中最高的下調基因(P=1.2×10?5,fold change=2.73;P值是用edgeR中的經驗Bayes準似然F檢驗計算的)。CD103通常高表達于腫瘤浸潤的Treg細胞上,并標志著一種特別具有抑制作用的Treg細胞亞群。因此,CD103在Pglrp1-/-Treg細胞上的異常表達可能與其在腫瘤中的低表達有關。
Pglyrp1–/–和WT小鼠之間的CD8+ T細胞特征更加不同,有更多的差異表達基因(239個上調和226個下調;圖3c),在主成分(PC)空間中距離更大(最明顯在干細胞簇中;圖3c),并且有一個明顯的全局偏移(圖3e)。此外,效應器/耗竭細胞的比例在Pglyrp1–/– TILs中顯著增加(圖3d),與先前的流式分析一致(圖2e–g)。這些數據表明干細胞簇中存在重大表達變化,以及Pglyrp1–/–小鼠中效應器/耗竭細胞比例的增加,表明干細胞樣細胞向效應器/耗竭細胞的分化軌跡發生了變化。事實上,Pglyrp1–/–小鼠的干細胞樣細胞顯示出更高的基因(Stat1、Cd69、Jak2、Tbx21、Irf1和Prf1)和通路(IFN信號傳導、白細胞介導的細胞毒作用和耗竭)的表達水平,這些基因和通路參與了T細胞激活和效應T細胞的生成(圖3f,g)。這些數據進一步表明了CD8+ TILs中干細胞樣細胞向激活/耗竭細胞的轉變。
為了檢查PGLYRP1在CD8+ T細胞中是否具有細胞內功能,我們生成了有條件的Pglyrp1敲除小鼠(Pglyrp1fl/fl)并將它們與E8iCre小鼠交配,以生成CD8+ T細胞中特異性刪除Pglyrp1的小鼠(E8iCrePglyrp1fl/fl)。我們將MC38-OVA腫瘤植入E8iCrePglyrp1fl/fl小鼠和野生型同窩對照動物,并發現E8iCrePglyrp1fl/fl小鼠的腫瘤生長控制更好,并且CD8+ TILs中PD-1和TIGIT表達水平更高的細胞比例增加(圖3h,i)。相比之下,特異性在髓系細胞中刪除Pglyrp1表達(LysMCrePglyrp1fl/fl小鼠)并沒有改變腫瘤生長,這表明Pglyrp1在髓系細胞上的表達對抗腫瘤免疫是不必要的。
總之,我們的數據表明,在CD8+ TILs中刪除Pglyrp1導致干細胞樣細胞向激活/耗竭細胞的轉變,表明PGLYRP1可能是一個調節CD8+ TILs中從干細胞樣細胞到效應器的過渡的抑制分子。

5、PGLYRP1缺陷小鼠可免受EAE的侵襲
阻斷共抑制分子用于癌癥治療的一個主要障礙是誘導自身免疫樣副作用,這些副作用是由于免疫抑制分子在調節免疫反應和恢復體內平衡方面的重要作用而產生的。類似地,在小鼠模型中,阻斷和刪除T細胞抑制分子,如CTLA-4、PD-1、TIM-3和TIGIT,會導致加重的自身免疫疾病,包括EAE。由于小鼠的腫瘤模型持續時間較短,不允許我們研究自發性自身免疫,因此我們測試了Pglyrp1喪失對自身免疫模型的影響。
為了確定PGLYRP1缺乏是否也會加重自身免疫疾病,我們在Pglyrp1–/–小鼠和匹配的野生型同窩對照小鼠中誘導了EAE。令人驚訝的是,Pglyrp1–/–小鼠在EAE中獲得了高度的保護,發病率減少(Pglyrp1–/–:62%;WT:100%),最大臨床EAE評分(Pglyrp1–/–:2.3;WT:2.9)和平均臨床EAE評分(Pglyrp1–/–:1.6;WT:2.3;圖4a–c)。組織學分析進一步顯示,Pglyrp1–/–小鼠的中樞神經系統病變和視神經炎減少(圖4d,e)。與WT相比,在Pglyrp1–/–小鼠的中樞神經系統中,我們發現造血浸潤物(CD45+)以及CD4+ T細胞、CD8+ T細胞、Treg細胞和B細胞浸潤物普遍減少,這些細胞類型已知在EAE疾病中發揮重要作用(圖4f)。滲透到Pglyrp1–/–小鼠中樞神經系統的CD4+ T細胞表達的炎癥因子IL-17A、IFNγ和GM-CSF的水平以及抗炎因子IL-10顯著降低(圖4g)。滲入的CD8+ T細胞表達的IL-17A和IFNγ水平顯著降低。總之,這些數據表明在EAE期間沒有PGLYRP1的T細胞啟動缺陷。
總之,與其他已知的T細胞抑制分子缺陷的小鼠不同,Pglyrp1–/–小鼠對EAE獲得了保護,PGLYRP1似乎對EAE疾病病理起作用。

6、PGLYRP1在髓系細胞中作為促炎分子發揮作用
為了揭示全局Pglyrp1敲除小鼠(Pglyrp1–/–)中減少的EAE表型是由哪種細胞類型負責的,我們使用E8iCrePglyrp1fl/fl小鼠,并將有條件的Pglyrp1敲除小鼠(Pglyrp1fl/fl)與Cd4Cre小鼠交配,以生成CD4+ Tconv細胞、Treg細胞和CD8+ T細胞中Pglyrp1特異性刪除的小鼠(Cd4CrePglyrp1fl/fl)。與WT同窩小鼠相比,我們在這兩個模型中都沒有觀察到EAE的發展有明顯差異,這表明T細胞中PGLYRP1表達的喪失并不是Pglyrp1–/–小鼠中保護性EAE表型的原因(圖5a,b)。
基于在EAE期間Pglyrp1–/–小鼠中T細胞啟動缺陷(圖4f,g)和在EAE期間調節自反應T細胞啟動的髓系細胞的重要功能,我們假設破壞髓系細胞上PGLYRP1的表達可能有助于Pglyrp1–/–小鼠中EAE表型的減少。因此,我們將Pglyrp1fl/fl小鼠與LysMCre小鼠交配,以生成具有髓系細胞特異性Pglyrp1刪除的小鼠,包括單核細胞、成熟巨噬細胞、粒細胞和一些樹突細胞(DCs;LysMCrePglyrp1fl/fl)。有趣的是,與WT同窩對照小鼠相比,LysMCrePglyrp1fl/fl小鼠對EAE獲得了保護(圖5c,d),體內的炎癥因子IL-6水平較低(圖5e)。中樞神經系統內浸潤的髓系細胞表現較不活化,如主要組織相容性復合體II類(MHC II類)的表達(圖5f)。與減少的髓系細胞活化相一致,dLNs中的CD4+ T細胞似乎啟動較少,CD44激活標記物和細胞因子GM-CSF、IL-2和TNF的表達減少(圖5g),這表明LysMCrePglyrp1fl/fl小鼠在EAE期間T細胞啟動存在缺陷。此外,在中樞神經系統中檢測到較低頻率的致病性產生IL-17的輔助T(TH17)細胞(IL-17A+ IFNγ+;圖5h)。這些數據共同表明,在EAE期間,髓系細胞中的PGLYRP1表達是足夠的抗原呈遞和T細胞啟動所必需的。
為了檢驗PGLYRP1在髓系細胞中在抗原呈遞給CD4+ T細胞中的潛在作用,我們進行了抗原呈遞實驗。我們將來自LysMCrePglyrp1fl/fl小鼠或WT同窩的脾細胞與具有特異于髓鞘膠質細胞糖蛋白(MOG)的T細胞抗原受體(TCR)的2D2 TCR轉基因小鼠的初級CD4+ T細胞在培養基中進行共培養,其中包含MOG肽或不包含MOG肽。在具有LysMCrePglyrp1fl/fl脾細胞的培養孔中,2D2細胞的增殖率較低,并且較低水平的促炎細胞因子IL-1α、IL-6、MCP-1和TNF分泌到培養基中,表明PGLYRP1在髓系細胞中在抗原呈遞和CD4+ T細胞啟動中起作用。
為了檢驗PGLYRP1在髓系細胞中是否在EAE期間的T細胞啟動中發揮作用,我們在誘導EAE的背景下在LysMCrePglyrp1fl/fl小鼠和WT同窩對照小鼠中進行了一次召回實驗。在免疫后的第10天,我們收集脾臟并將脾細胞與或不與MOG肽一同培養。在具有LysMCrePglyrp1fl/fl脾細胞的培養孔中,培養基中檢測到較低濃度的促炎細胞因子(IFNβ、IL-6、MCP-1和TNF),這表明PGLYRP1在髓系細胞中在EAE期間CD4+ T細胞啟動中起作用。
綜上所述,這些數據表明,在EAE期間,髓系細胞需要PGLYRP1進行最佳的抗原呈遞和CD4+ T細胞的啟動。

7、Plyrp1-/-單核細胞和中性粒細胞的表達變化
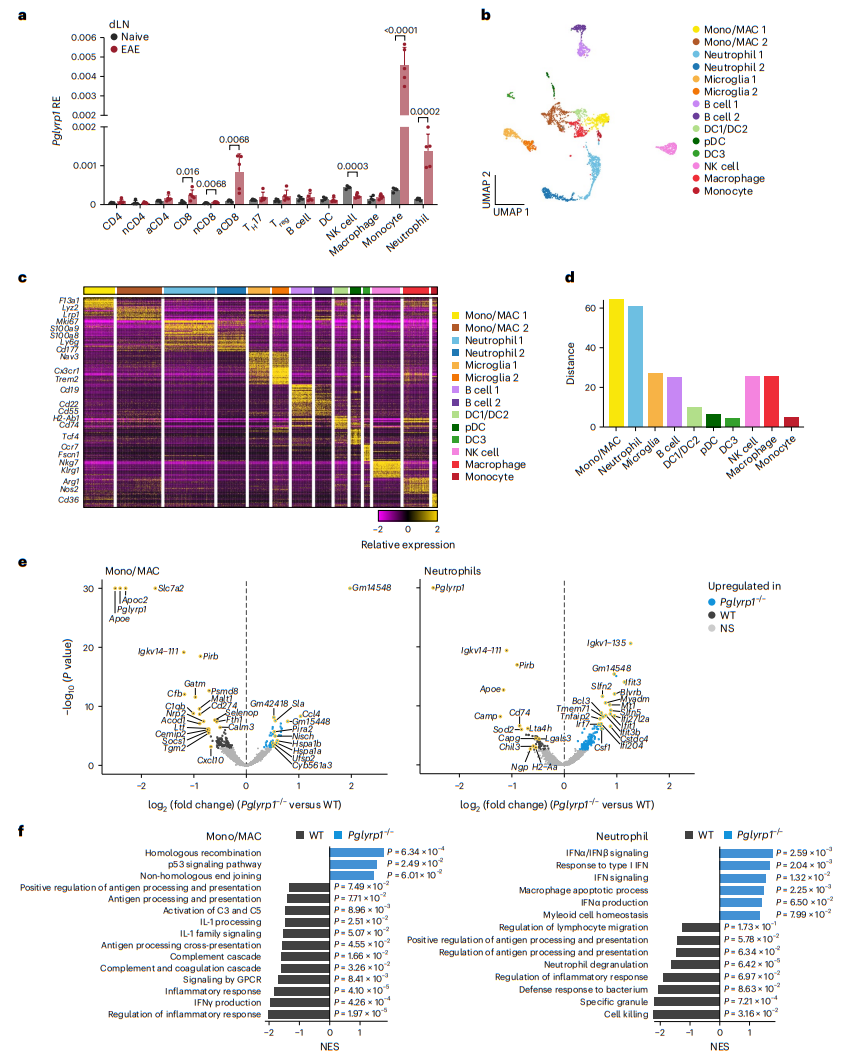
為了揭示在EAE期間PGLYRP1缺陷影響哪些髓系細胞群,我們首先分析了在EAE發作初期的淋巴結內的免疫群體中Pglyrp1的表達情況(圖6a)。我們檢測到單核細胞和中性粒細胞中表達最高。這種高Pglyrp1表達是由EAE誘導的,因為來自天然小鼠的單核細胞和中性粒細胞顯示出較低的Pglyrp1表達。
為了檢驗PGLYRP1缺陷如何改變髓系細胞組分,我們對來自Pglyrp1–/–小鼠和WT對照小鼠的EAE初發時的CD45+中樞神經系統浸潤細胞進行了單細胞RNA測序,并分析了B細胞和髓系細胞(CD3–細胞)。3698個主要是髓系細胞的細胞譜被分為14個簇(圖6b),具有明顯的表達特征(圖6c)。我們使用細胞類型標記和差異基因簽名將這14個亞群進行了注釋,包括單核細胞/巨噬細胞、中性粒細胞、小膠質細胞、B細胞、DC1s/DC2s、漿細胞樣DC、DC3s、NK細胞、巨噬細胞和單核細胞(圖6c)。
在每個簇中,Pglyrp1–/–和WT細胞之間的差異(主成分空間中的距離)最大(圖6d)。在Pglyrp1–/–和WT小鼠之間的不同基因表達分析中,單核細胞/巨噬細胞(44個基因上調和119個基因下調)和中性粒細胞(163個基因上調和54個基因下調)之間的差異最大(FDR < 0.05且|log2(fold change)| > 0.25;FDR是通過與單核細胞/巨噬細胞相同的方法計算的;圖6e)。在Pglyrp1–/–小鼠中,單核細胞/巨噬細胞中減少的基因包括互補系統成員(Cfb和C1qb)和促炎趨化因子CXCL10(Cxcl10)的編碼基因。Pglyrp1–/–中性粒細胞中Cd74的表達,該基因編碼HLA-DR抗原相關不變鏈(CD74),較低。有趣的是,Pglyrp1–/–單核細胞/巨噬細胞和中性粒細胞中有九個基因下調,包括Apoe和Lgals3。特別是,在Pglyrp1–/–單核細胞/巨噬細胞(變化排名第1)和中性粒細胞(變化排名第3)中,Apoe顯著下調。Apoe編碼載脂蛋白E,最近已經顯示它在髓系細胞抗原呈遞和T細胞啟動中具有關鍵作用,與我們在EAE期間的Pglyrp1–/–小鼠中發現的抗原呈遞和T細胞啟動的缺陷一致(圖4g和圖5g、i)。Lgals3編碼加凝集素-3,調節單核細胞/巨噬細胞的遷移。在Pglyrp1–/–單核細胞/巨噬細胞和中性粒細胞中,多個與髓系細胞激活有關的基因上調,包括Jun和Lilra6,它們都參與髓系細胞激活。Pglyrp1–/–中性粒細胞中上調的多個干擾素刺激基因(Ifit3、Ifit3b、Ifit1、Ifi204、Ifi27l2a、Oasl2和Irf7)表明了IFN信號的增加。WT單核細胞/巨噬細胞中不同上調的基因富集了互補級聯、炎癥反應和抗原呈遞通路,而Pglyrp1–/–單核細胞/巨噬細胞上調的基因富集了DNA修復和細胞分裂通路(圖6f)。WT中性粒細胞富集了涉及殺傷細胞、中性粒細胞顆粒釋放和抗原呈遞的通路,而Pglyrp1–/–中性粒細胞中富集了與干擾素信號有關的通路。綜上所述,這些分析表明,在Pglyrp1–/–單核細胞/巨噬細胞和中性粒細胞中減少的促炎通路和激活,以及巨噬細胞的抗原呈遞。
PGN與PGLYRP1結合是其通過TREM-1激活髓系細胞的能力所必需的。為了測試PGLYRP1本身是否也能激活髓系細胞,我們在體外用PGN、重組PGLYRP1或PGN + 重組PGLYRP1處理單核細胞,并測量促炎細胞因子TNF分泌到培養基中。我們發現只有經過PGN和PGLYRP1處理的單核細胞顯著分泌TNF,這表明與PGN結合的PGLYRP1介導髓系細胞的激活,在沒有PGLYRP1的情況下,髓系細胞不能有效地被激活,從而無法在EAE期間進行最佳的抗原呈遞。
總之,我們發現單核細胞和中性粒細胞中Pglyrp1的高表達以及在EAE期間Pglyrp1–/–小鼠中這些細胞群體中出現大量的轉錄變化。這些數據支持這樣一個模型,即在單核細胞/巨噬細胞和中性粒細胞中PGLYRP1的表達對于EAE中致病反應的最佳啟動是必需的。單核細胞/巨噬細胞和中性粒細胞的抗原呈遞所需功能的缺陷進一步導致了Pglyrp1–/–小鼠和LysMCrePglyrp1fl/fl小鼠EAE疾病的減輕。
實驗方法
初代CD4+T細胞的分離與分化、主動誘導EAE、CNS 組織學、腫瘤實驗、流式細胞術、熒光激活細胞分選、抗原呈遞試驗、召回實驗、基于微珠的免疫分析、qPCR、Bulk RNA-seq、scRNA-seq、細胞類型注釋、RNA速度分析、GSEA。
參考文獻
Schnell, A., Huang, L., Regan, B.M.L. et al. Targeting PGLYRP1 promotes antitumor immunity while inhibiting autoimmune neuroinflammation. Nat Immunol (2023).