乳酸化修飾研究摘要匯總
乳酸化是一種新的乳酸衍生的組蛋白翻譯后修飾(PTM),是由芝加哥大學趙英明教授課題組首先發現的,該研究揭示了乳酸在表觀遺傳學途徑中的作用,首次報道乳酸可以修飾組蛋白,調控基因的轉錄表達,拓展了大家對乳酸的認識。
乳酸(lactate)是細胞糖酵解途徑重要的含碳代謝產物,已成為一種能量底物和有價值的信號分子。細胞代謝過程中,乳酸的積累促進了組蛋白賴氨酸發生乳酸化修飾。乳酸化修飾來源于細胞葡萄糖代謝產生的乳酸,并受到糖酵解和線粒體氧化代謝的調控。
由于 Warburg 效應(有氧糖酵解)是癌癥的標志之一,即使在有氧條件下,癌細胞也傾向于將葡萄糖轉化成乳酸來產生能量,與正常細胞相比,葡萄糖代謝的乳酸更多。因此,腫瘤中的組蛋白乳酸化很可能是異常的。
乳酸的生物學功能也因腫瘤細胞中Warburg效應的存在,得到了廣泛關注。
相關最近研究文章
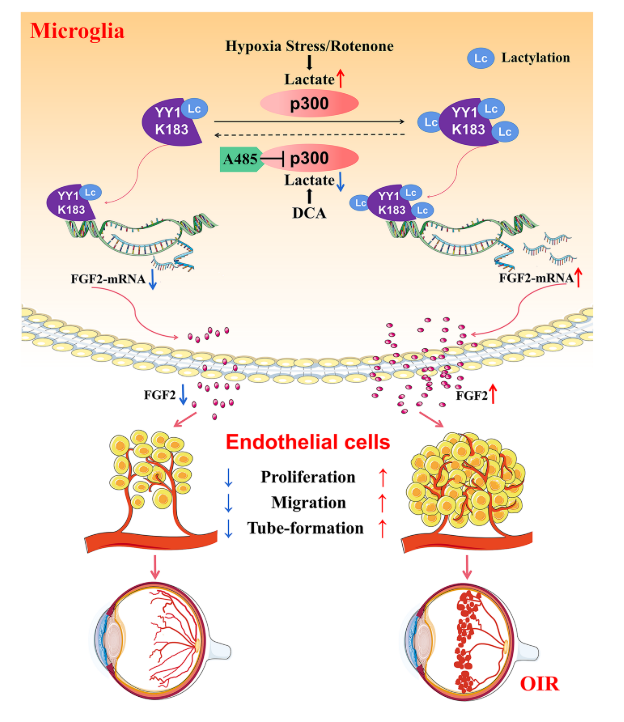
1.小膠質細胞中YY1乳酸化通過轉錄激活介導的FGF2上調促進血管生成

摘要
背景:眼部新生血管是致盲的主要原因。視網膜小膠質細胞與缺氧誘導的血管生成和血管病變有關,但其潛在機制尚不完全清楚。乳酸化是一種新的乳酸衍生的翻譯后修飾,在多種細胞過程中起關鍵作用。缺血性視網膜病變缺氧是視網膜新生血管形成的促發因素,因此乳酸化很可能參與了這一過程。本研究旨在探討乳酸化在視網膜新生血管形成中的作用,并為視網膜新生血管疾病尋找新的治療靶點。
結果:通過集落刺激因子1受體(CSF1R)抑制劑PLX 3397去除小膠質細胞抑制氧誘導的視網膜病變中的視網膜新生血管形成。缺氧增加小膠質細胞中的乳酸化并加速FGF2表達,促進視網膜新生血管形成。確定了67個蛋白質的77個位點在缺氧條件下乳酸增加的背景下具有增加的乳酸化。研究結果表明,轉錄因子非組蛋白Yin Yang-1(YY 1)在第183位賴氨酸(K183)發生了乳酸化,該位點受組蛋白乙酰轉移酶p300的調控。高乳酸化YY 1直接增強FGF2轉錄并促進血管生成。K183的YY1突變消除了這些影響。p300的過表達增加YY1乳糖化并增強體外血管生成,并且施用p300抑制劑A485極大地抑制體內和體外血管生成。
結論:上述研究結果表明,YY1乳糖化在小膠質細胞中起著重要的作用,通過上調FGF2的表達在視網膜新生血管。靶向乳酸/p300/YY1乳酸化/FGF2軸可為增殖性視網膜病提供新的治療靶標。
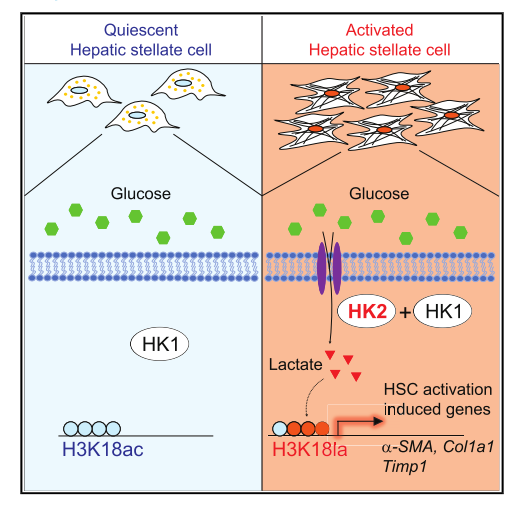
2.己糖激酶2通過組蛋白乳酸化介導的基因表達是肝星狀細胞活化和肝纖維化所必需的

摘要
乳酸與肝星狀細胞(HSC)的活化有關。然而,乳酸發揮其作用的機制仍然難以捉摸。使用RNA-seq和CUT&Tag染色質分析,我們發現在活化的HSC中誘導己糖激酶2(HK 2)表達是通過組蛋白乳酸化而不是組蛋白乙酰化。通過Hk2缺失抑制組蛋白乳酸化或乳酸鹽產生的藥理學抑制減少HSC活化,而外源性乳酸鹽而非乙酸鹽補充挽救活化表型。因此,由活化的HSC產生的乳酸鹽通過組蛋白乳酸化決定HSC的命運。研究發現組蛋白乙酰化與組蛋白乳酸化競爭,這可以解釋為什么I類HDAC(組蛋白脫乙酰酶)抑制劑阻礙HSC活化。最后,HSC特異性或全身性缺失HK2抑制HSC活化和體內肝纖維化。因此,本研究提供證據表明HK2可能是肝纖維化的有效治療靶點。
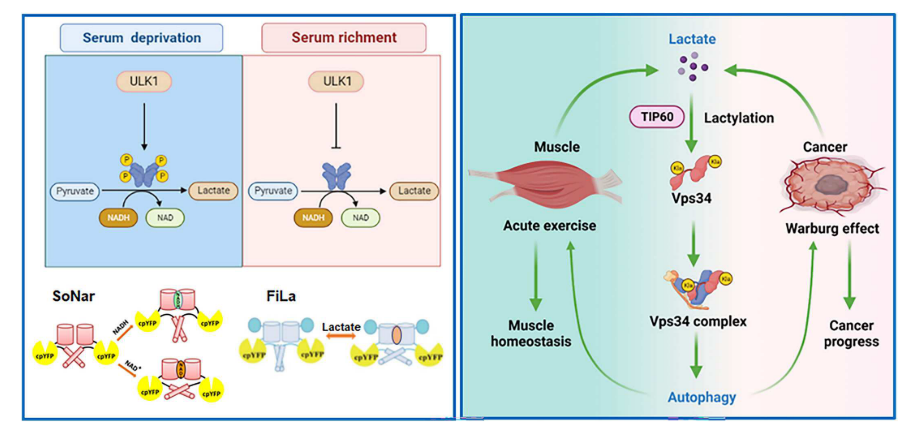
3.ULK1介導的代謝重編程通過其乳酸化調節Vps34脂質激酶活性

摘要
自噬和糖酵解是涉及生理和病理細胞程序的高度保守的生物過程,但這些過程之間的相互作用知之甚少。在這里,表明了糖酵解酶乳酸脫氫酶A(LDHA)被激活后UNC-51樣激酶1(ULK1)激活營養剝奪。具體而言,ULK 1直接與LDHA相互作用,在營養缺乏時磷酸化絲氨酸-196并促進乳酸產生。乳酸通過酰基轉移酶KAT5/TIP 60介導Vps34乳酸化(在賴氨酸-356和賴氨酸-781處),從而連接自噬和糖酵解。Vps 34乳酸化增強了Vps34與Beclin1、Atg14 L和UVRAG的結合,然后增加了Vps34脂質激酶活性。Vps34乳酸化促進自噬通量和內溶酶體運輸。劇烈運動期間骨骼肌中的Vps 34乳酸化維持肌細胞穩態,并通過誘導細胞自噬與癌癥進展相關。本研究結果描述了自噬的調控機制,并將細胞自噬和糖酵解結合起來。
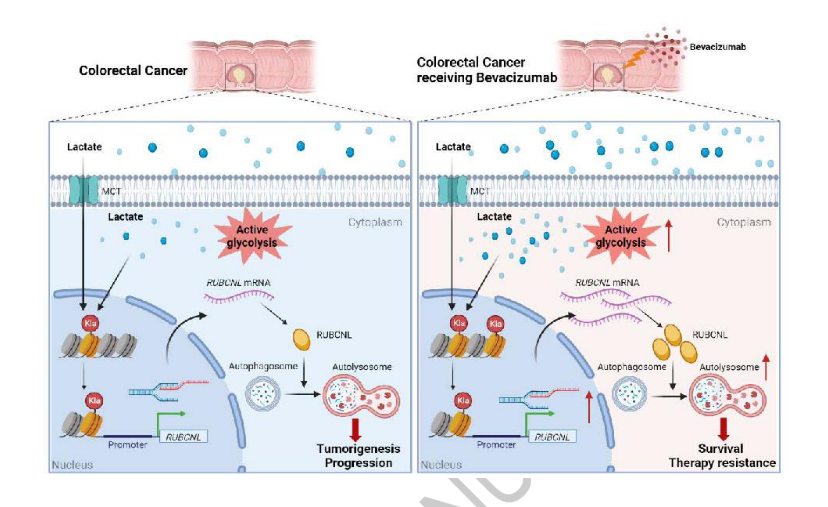
4.腫瘤來源的乳酸鹽通過組蛋白H3賴氨酸18乳酰化(H3K18la)促進結直腸癌中自噬增強蛋白RUBCNL表達來促進對貝伐單抗治療的抗性

摘要
貝伐單抗在轉移性結直腸癌(CRC)的一線和二線治療中起重要作用。缺氧的誘導和腫瘤對缺氧的反應在決定抗血管生成治療的療效方面起著重要的作用,但兩者之間的聯系尚不清楚。在這里,發現乳酸積累在腫瘤環境的CRC和作為底物的組蛋白乳酸酰化,這一過程進一步誘導細胞增強糖酵解缺氧。對貝伐單抗治療具有抗性的CRC患者呈現升高的組蛋白乳酸化水平,并且組蛋白乳酸化的抑制有效地抑制CRC腫瘤發生、進展和在缺氧中的存活。組蛋白乳酸化促進RUBCNL/Pacer的轉錄,通過與BECN 1(beclin 1)相互作用促進自噬體成熟,并介導III類磷脂酰肌醇3-激酶復合物的募集和功能,其在缺氧癌細胞增殖和存活中起關鍵作用。此外,在貝伐珠單抗抗性患者衍生的臨床前模型中,將組蛋白乳酸化和巨自噬/自噬的抑制與貝伐珠單抗治療組合顯示出顯著的治療功效。這些發現為代謝重編程-表觀遺傳調控提供了新的探索和重要補充,并為通過抑制組蛋白乳酸化提高貝伐珠單抗在結直腸癌中的臨床療效提供了新的策略。
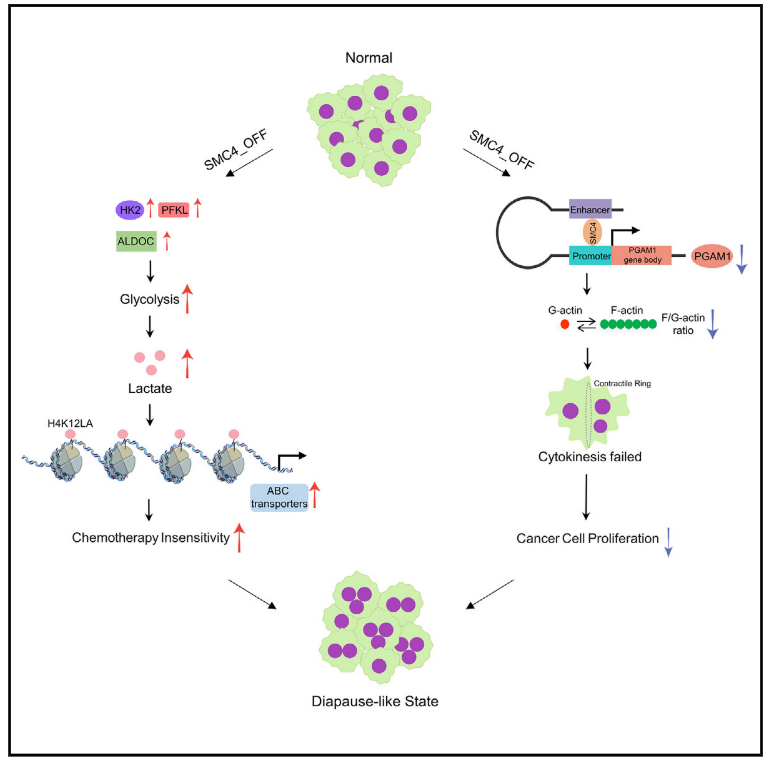
5.SMC4減少誘導的滯育樣結直腸癌細胞具有低增殖和化療不敏感的特征

摘要
為了應對不利的環境條件,胚胎發育可能會可逆地停止,這一過程稱為滯育。最近的報道將這一現象與腫瘤對化療的非遺傳反應聯系起來,但所涉及的機制知之甚少。在這里,建立了4號染色體結構維持(SMC4)在結腸直腸癌細胞向滯育樣狀態轉換中的多種作用。SMC4減弱促進三種投資期糖酵解酶的表達,增加乳酸產生,同時還抑制磷酸甘油酸變位酶1(PGAM1)。所得的高乳酸水平通過組蛋白乳酰化增加ABC轉運蛋白表達,使得腫瘤細胞對化療不敏感。SMC 4充當PGAM 1轉錄的共激活因子,并且SMC4和PGAM 1的協調丟失影響F-肌動蛋白組裝,誘導胞質分裂失敗和多倍體,從而抑制細胞增殖。這些對非遺傳性化療耐藥性機制的見解可能對該領域具有重大意義,推進對腫瘤中有氧糖酵解功能的理解,并可能為未來的治療策略提供信息。