






mSWISNF復合物在TKIs耐藥EGFR突變肺癌中的調控機制

EGFR突變型肺腺癌可獲得酪氨酸激酶抑制劑(TKI)耐藥,比如奧西替尼,這通常是由非遺傳機制引起。作者鑒定了奧西替尼敏感和耐藥的EGFR突變細胞和患者腫瘤衍生模型中的染色質可及性變化和基因調控特征,并揭示了哺乳動物SWI/SNF染色質重塑復合物在TKI耐藥性中的作用。通過分析mSWI/SNF全基因組定位,作者確定了奧西替尼抗性狀態下癌細胞特異性基因靶點。重要的是,基因層面和藥理學層面破壞SMARCA4/SMARCA2 mSWI/SNF ATP酶,可通過抑制mSWI/SNF介導的細胞程序調節細胞增殖、上皮-間充質轉化、上皮細胞分化和NRF2信號傳導,使一些耐藥模型對奧西替尼重新敏感。這些數據強調了mSWI/SNF復合物在TKI耐藥性中的作用,并表明mSWI/SNF抑制劑在TKI耐藥性肺癌中的潛在效果。該研究于2023年8月發表在 《Cancer Cell》,IF:50.3。
技術路線

主要研究結果
1. 奧西替尼耐藥的EGFR突變癌細胞發生染色質可及性變化
為研究EGFR突變癌細胞奧西替尼的耐藥性機制,作者產生了五對親代和奧系替尼耐藥性的EGFR突變癌細胞系(圖1A)。親本細胞系對奧西替尼的EC50為10 nM或更低,而其抗性細胞系EC50高出>90倍(圖1B和1C)。然后對這些細胞系對進行RNA-seq,確定抗性細胞的基因調控譜。作者鑒定到了不同的上調和下調基因,其中許多差異基因在至少2個細胞系之間共有(圖1D)。C3和C8分別包含五種奧西替尼抗性而非吉非替尼抗性狀態中的下調和上調基因,包括MMP2和FGF1(下調)以及ZEB2、ATF3、ETS-1和FYN(上調)等基因。作者在一個抗性細胞系中鑒定到許多獨特的差異調節基因,強調了抗性狀態的異質性(圖1D)。EMT和炎癥反應等通路在某些細胞系中上調,而MYC靶點、干擾素α/γ反應信號通路則顯示下調(圖1E)。
接下來,作者在四個細胞系對進行ATAC-seq,其中包括PC9*、HCC406*、HCC827*和PC9(圖1F)。作者鑒定到抗性細胞中抗性相關的可及性基因組差異位點(圖1F)。將這些數據與RNA-seq數據結合,發現>50%基因的表達變化與基因啟動子或增強子處或附近的DNA可及性變化一致(圖1G、1H)。這些發現證實EGFR突變NSCLC癌癥細胞系中抗性和親代狀態之間的染色質可及性變化是表征TKI抗性基因調控程序的基礎。

圖1:EGFR突變肺癌細胞中奧西替尼耐藥的染色質可及性和基因調控基礎
2. mSWI/SNF復合物是奧西替尼耐藥基因程序和染色質可及性的上游調節因子
染色質可及性變化是TKI抗性的一個關鍵基因調控特征,接下來作者試圖預測控制這些變化的潛在染色質相關調控因子。在所有細胞系中,對奧西替尼抗性(OR)(或吉非替尼抗性)狀態下的差異基因表達譜(RNA-seq)進行IPA分析,mSWI/SNF復合物的ATP酶亞基SMARCA4關聯度最高(圖2A)。其他包轉錄因子(TF)括,如TP63、STAT3和SOX2,其中一些通過mSWI/SNF復合物以及ARID1A與染色質相關連,ARID1A是典型BAF(cBAF)mSWI/SFF亞復合物亞基(圖2A)。
為了解SMARCA4如何調節基因表達,作者使用CUT&RUN分析PC9*/OR*、HCC406/OR和HCC827*/GR6細胞系中mSWI/SNF復合物全基因組的占有情況(圖2B)。將這些數據與ATAC-seq進行整合,確定了在抗性狀態下獲得或丟失的位點子集(圖2C)。獲得和丟失位點主要位于啟動子遠端,并且在AP1家族基序上富集(圖2B、2C)。對這些基因進行排序,其在抗性狀態下mSWI/SNF特異性結合與可及性變化表現一致(圖2D)。EMT(TWIST1和ZEB2)、細胞遷移(MMP13、BMP4和COL4A1)和RTK信號傳導基因上調,而編碼上皮細胞分化和信號傳導(FGFR1/2、JAG1和NOTCH1)的基因在抗性狀態下下調。參與MAPK信號傳導的基因表達(MAPK1、DUSP6和SPRY4)以細胞系特異性的方式發生改變(圖2D)。親本和抗性狀態之間mSWI/SNF占有率、可及性和基因表達的一致變化發生在ETV1和TWIST1基因座(圖2E)。這些發現表明,在TKI抗性EGFR突變細胞系中,mSWI/SNF復合物在抗性相關基因座上的活性起著關鍵作用。
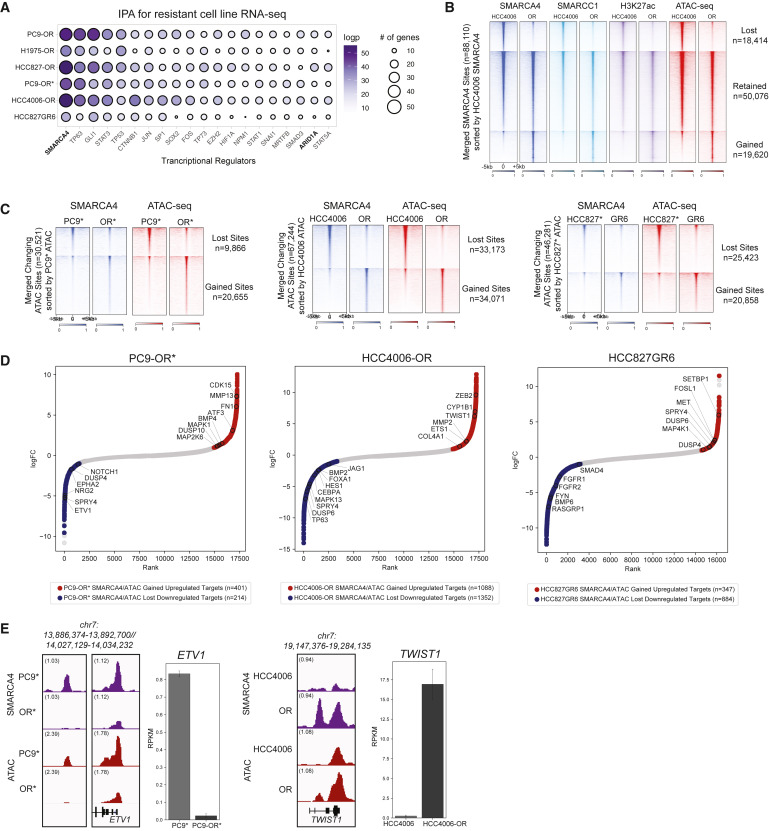
圖2:哺乳動物SWI/SNF(BAF)復合物是抗性相關基因的關鍵調控因子
3.抑制SMARCA4使奧西替尼耐藥模型對奧西替尼重新敏感
作者在PC9/PC9-OR、H1975/H1975-OR和HCC827/HCC827-OR親本和抗性細胞系中進行shRNA介導的SMARCA4敲除(圖3A-3D)。與H1975-OR和HCC827-OR細胞相比,SMARCA4敲低對奧西替尼處理后的PC9-OR細胞影響最顯著。與未處理細胞相比,奧西替尼的存在使PC9-OR細胞增殖完全消除(圖3B和3C)。此外, SMARCA4敲低后奧西替尼EC50降低了8倍(圖3D)。
同時,作者利用對奧西替尼具有敏感性的EGFR突變肺癌的患者衍生異種移植物(PDX)來觀察SMARCA4敲低的影響。通過免疫組織化學(IHC)檢查這些PDX中SMARCA4的水平,發現所有腫瘤都產生SMARCA4(圖3E)。因為YU-005X對奧西替尼表現出原發性耐藥性,作者使用源自該腫瘤的細胞系(YU-005C細胞)。SMARCA4敲除顯著損害YU-005C細胞增殖和形成集落的能力,而添加奧西替尼則加劇了這種效果(圖3F和3G)。體內實驗中,作者在YU-005C細胞來源的腫瘤中敲除SMARCA4(圖3H和3L)。皮下注射YU-005C細胞導致腫瘤形成,SMARCA4基因敲除與多西環素處理后腫瘤繼續生長(圖3H)。SMARCA4基因敲除和處理則抑制腫瘤(圖3H和3I),而對照SMARCA4野生型腫瘤生長沒有受到奧西替尼處理的影響。這些結果表明,奧西替尼耐藥腫瘤在奧西替尼存在下依賴SMARCA4進行增殖。
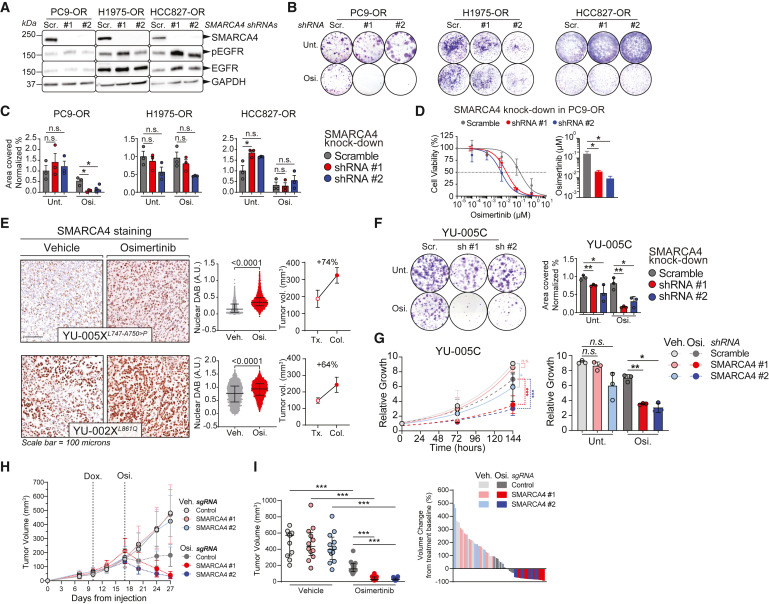
圖3: SMARCA4缺失使一部分耐藥腫瘤對奧西替尼敏感
4. SMARCA4丟失改變染色質可及性、抗性和增殖相關基因譜表達
為研究SMARCA4如何在奧西替尼存在的情況下維持癌癥細胞,作者在PC9-OR和YU-005C細胞中在奧西替尼存在或不存在的條件下敲低SMARCA4,然后進行RNA-seq和染色質可及性(ATAC-seq)分析(圖4A)。兩種細胞中均檢測到上調和下調基因,與對照相比,并確定了奧西替尼處理后SMARCA4缺失時其表達逆轉的top基因,因為在這些條件下觀察到不同的細胞活力表型(圖4B和4C)。在PC9-OR和YU-005C細胞中,SMARCA4敲低引起的上調和下調基因在特定信號通路和細胞增殖和免疫信號相關的生物進程中富集(圖4D)。此外,在PC9-OR細胞的耐藥性相關DEG中,發現在SMARCA4敲低和奧西替尼處理后,20%的DEG被解除調節(圖4E),其中包括分別參與細胞存活和異生代謝過程的基因,如BCL2或CYP26a1(圖4E)。聯合ATAC-seq,在PC9-OR和YU-005C細胞中,SMARCA4抑制和奧西替尼處理后,鑒定到了可及性和基因表達均降低的關鍵基因(圖4F和4G)。一個子集屬于AP-1 TFs家族(FOS和JUNB)以及參與癌癥和增殖的其他基因,如HES1、EGR1和JAG1(圖4G)。這些數據強調了SMARCA4缺失對維持抗性狀態的影響,并確定了受其破壞影響的關鍵基因。
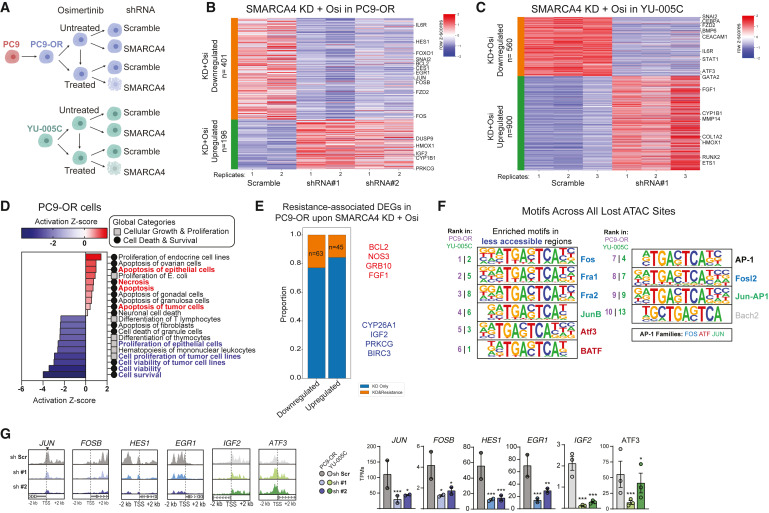
圖4:奧西替尼耐藥細胞系中SMARCA4抑制可調節耐藥相關基因
5.藥理學抑制mSWI/SNF ATP酶活性逆轉EGFR突變癌細胞對奧西替尼的抗性
Cmp14是特異性變構SMARCA4/2 ATP酶的抑制劑。作者使用combenefit測定法測試幾個藥物協同作用(圖5A)。作者先前的工作表明ERK再激活是EGFR-TKI治療失敗的主要決定因素,通過與MEK抑制劑聯合治療可以避免這種情況。在Cmp14存在或不存在的條件下,用奧西替尼和曲美替尼(OT)處理PC9*和PC9-OR*細胞,用于組合測定(圖5A)。
combenefit分析揭示了OT和Cmp14之間的顯著協同作用,其對PC9-OR*細胞具有特異性(圖5A)。與單獨OT處理相比,Cmp14額外添加增強了PC9-OR*中的細胞凋亡(圖5B)。為確定在PC9-OR*細胞中觀察到的藥物協同作用背后的轉錄和染色質可及性變化,作者進行了RNA-seq和ATAC-seq實驗(圖5C)。對DMSO、OT和OT+Cmp14之間差異調節基因進行聚類分析,其結果顯示四組關鍵基因(圖5D)。C1包括DMSO組與Osi+Tram組中下調而在OT+Cmp14組中上調的基因, C2包含在OT處理激活但在OT+Cmp14時強烈下調的基因,C3包括其表達因OT處理而輕度降低,但因OT+Cmp14處理而強烈降低的基因, C4包括在對照和OT處理條件下都有活性,但與Cmp14聯合后強烈下調的基因(圖5D)。這些數據與ATAC-seq結合后揭示了與這四個聚類中基因相對應的基因座染色質可及性與基因表達變化相關(圖5D)。接下來,作者對C1-C4轉錄反應進行表征,突出其“主要”靶標(具有與染色質可及性變化一致的靶標)和“次要”靶標(缺乏可及性一致變化的靶標)(圖5E)。OT+Cmp14處理后下調的基因包括參與細胞周期和凋亡、細胞遷移和粘附、代謝過程和核受體通路因子,包括NRF2信號和毒素代謝,如EPHA4,RAC2,CDC25C和GPX2(圖5D-5F)。上調基因(C1)包括參與蛋白激酶活性、免疫信號傳導和核受體(如ATF3和CYP1B1)等過程基因(圖5E和5F)。在8344個對OT+Cmp14表現出可及性降低的位點中,發現超過40%的位點在PC9-OR*細胞中含有BAF復合物峰(圖5G,頂部)。此外,這些位點的一個子集(綠色圓圈)被定位到抗性狀態下選擇性上調但僅在OT+Cmp14聯合處理時下調的基因(品紅色圓圈)(圖5G,底部)。這在CES1基因座上得到例證,在該基因座上,作者觀察到在OR*(耐藥)環境中BAF復合體的占有率和可及性增加,而在OT+Cmp14治療后,這種情況顯著降低(圖5H)。CES1是一種關鍵的NRF2調節酶,它介導外源性代謝,并與肝細胞癌的化療耐藥性有關。
最后,作者旨在確定抵抗狀態下only上調或下調的基因(即PC9-or*與PC9*)是否可以在OT+Cmp14聯合治療環境中相對于僅OT選擇性逆轉。在鑒定60個的基因中,其表達在抗性狀態下下調,單獨OT處理未能改變其表達,但OT+Cmp14聯合處理(TGFA、GDF15和NDRG1)后其表達逆轉(上調)(圖5I)。同時,在76個基因中, OT+Cmp14聯合處理(COL4A1、MAP2K6和EPHX1)中,其抗性相關表達被選擇性逆轉(下調)(圖5I)。此外,作者還分析了一組基因,這些基因在PC9*細胞經OT處理后表達發生變化(上調或下調的基因),而在PC9-OR*細胞中也做出相應反應(圖5J)。這些結果表明,抑制mSWI/SNF ATP酶與OT發生協同作用,部分通過重新聯系染色質可及性來逆轉耐藥狀態下的部分轉錄程序,使耐藥PC9-OR*細胞對TKI處理重新敏感。

圖5:靶向mSWI/SNF復合物ATP酶活性的藥理學逆轉EGFR突變癌細胞系亞群中的TKI耐藥性程序
6.由mSWI/SNF復合物降低的活性氧與奧西替尼重新敏感有關
接下來,作者試圖通過SMARCA4/2抑制或SMARCA4敲低來鑒定對奧西替尼重新致敏的EGFR突變細胞系潛在標志物或標志性特征。作者分析了對奧西替尼重新致敏的細胞系與未遵循SMARCA4 敲低或藥物抑制細胞系之間耐藥性DEG的差異。這揭示了PC9-OR(再敏化)細胞系與H1975-OR和HCC827-OR(未敏化)細胞株之間的差異,以及PC9-OR*(再敏化的)細胞系和HCC4006-OR(非敏化的)之間的差異(圖6A和6B)。GSEA通路分析顯示,相對于未致敏細胞系,再致敏細胞系PC9-OR和PC9-OR*中于PI3K信號傳導和活性氧(ROS)通路等通路富集(圖6B)。每個再致敏細胞系也表現出干擾素α/γ信號特異性陽性或陰性富集,表明細胞系特異性通路功能(圖6B)。同時,為確定可能增強PC9-OR和PC9-OR*細胞對奧西替尼或OT處理再致敏反應的基因靶點,作者鑒定了PC9-OR*和PC9-OR細胞與HCC406-OR、H1975-OR或HCC827-OR相比 的DEG,其中特異性上調和下調的DEG(圖6C)。某些DEG富集通路與MAPK信號傳導和解毒有關,包括上調基因MAPK1、CRKL和CES1,以及下調基因PLK3和ARNT2(圖6D)。
接下來,作者在PC9-OR*細胞、PC9-OR細胞和YU-005C細胞中研究與SMARCA4抑制或敲除后BAF介導的基因表達變化相關的染色質可及性變化基序(圖6E和6F)。PC9-OR*細胞中與Cmp14協同的基因附近可及性降低的位點中,與AP-1因子和NRF2相對應的基序是參與協同反應的假定候選者(圖6E),在PC9-OR和YU-005C細胞中SMARCA4敲低和奧西替尼處理后失去可及性位點的基序分析也顯示NRF2是一個候選因子(圖6F)。
NRF2信號通路負責在氧化應激條件下通過激活抗氧化反應清除活性氧。為驗證SMARCA4/2介導的奧西替尼重新致敏、ROS和解毒通路之間的相關性,作者研究了奧西替尼如何影響PC9-OR和YU-005C TKI抗性細胞中的ROS水平。奧西替尼處理后,PC9細胞中ROS水平顯著增加,這與NRF2信號整體減少一致(圖6G、6H)。相反,PC9-OR細胞中基線ROS顯著更高,奧西替尼沒有深刻影響ROS水平,這與活性NRF2信號一致(圖6G、6H)。在奧西替尼處理的PC9-OR細胞中敲低SMARCA4進一步增加ROS水平,同時降低NRF2通路活性,表明SMARCA4對于ROS中起著關鍵作用(圖6H和6I)。與這些觀察結果一致的是,SMARCA4敲除后,在奧西替尼治療的腫瘤中,YU-005C皮下腫瘤中的ROS水平最高(圖6J)。作者接下來研究NRF2在抗性表型中的功能。NRF2敲低降低了PC9-OR細胞對奧西替尼的敏感性,這證實NRF2是維持抗性的重要因子。此外,作者測試ROS存在細胞對奧西替尼反應中是否有直接影響。作者在EGFR突變腫瘤的組織微陣列中檢測NRF2和SMARCA4的水平。這些腫瘤中SMARCA4水平與核NRF2呈正相關,進一步支持這兩種蛋白質可能共同調節氧化應激(圖6L)。這些結果證實了SMARCA4染色質重塑活性在通過NRF2激活控制奧西替尼誘導的耐藥細胞氧化應激水平中的關鍵作用。
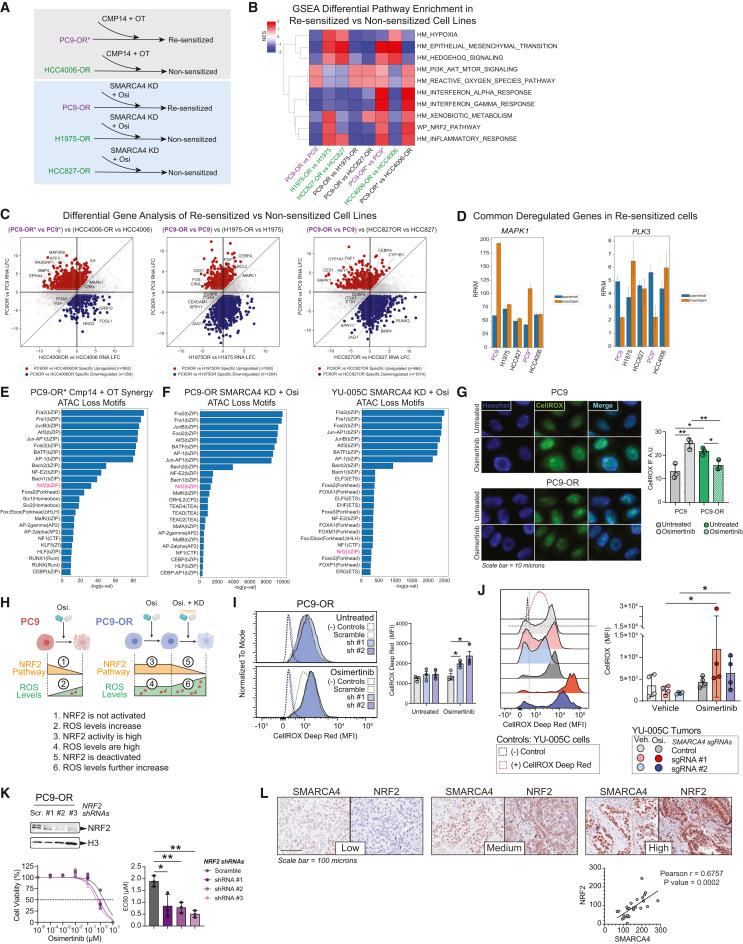
圖6:奧西替尼抗性細胞系減敏揭示了mSWI/SNF復合物降低活性氧
7.奧西替尼抗性PDX小鼠模型中藥理學抑制mSWI/SNF ATP酶活性降低腫瘤生長
接下來,作者探討mSWI/SNF ATP酶活性的藥理學抑制在SMARCA4依賴性YU-005患者衍生模型中的作用。YU005C細胞對單獨的Cmp14或FHD-286(臨床階段雙重SMARCA4/2=抑制劑)處理具有耐藥性。然而,Cmp14和FHD-286均使YU005C細胞對奧西替尼敏感(圖7A-7C)。因此,作者使用FHD-286在體內研究SMARCA4/2抑制與奧西替尼結合的潛力(圖7D)。用載體、奧西替尼、FHD-286或奧西替尼和FHD-286組合處理荷瘤小鼠18天。雖然每種藥物單獨使用導致適度的腫瘤生長減弱,但奧西替尼+-FHD-286聯合治療顯著抑制體內腫瘤生長(圖7E)。
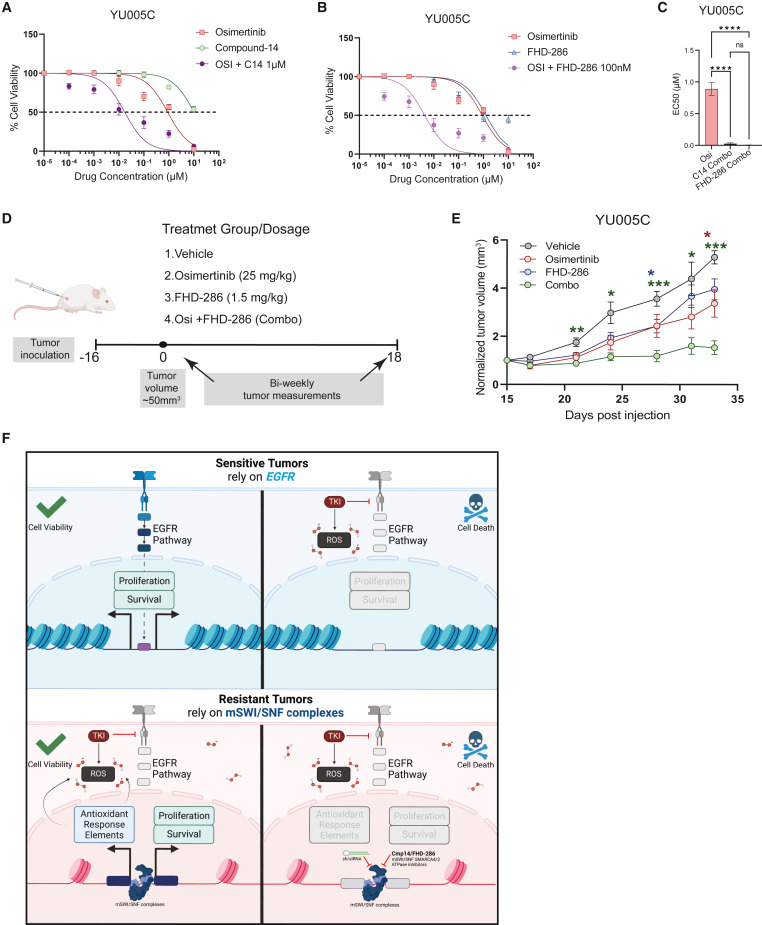
圖7:mSWI/SNSF ATP酶活性的藥理學抑制使患者來源的腫瘤對奧西替尼敏感
結論
基因敲低聯合抑制劑抑制mSWI/SNSF ATP酶活性可使TKI耐藥EGFR突變肺癌對奧西替尼重新敏感,并可增強細胞內活性氧水平且抑制腫瘤生長。
實驗方法
CUT&RUN,ATAC-seq,RNA-seq,IHC,IF,細胞培養,藥物實驗,細胞凋亡檢測,活性氧檢測,細胞活力檢測
參考文獻
de Miguel FJ, Gentile C, Feng WW, Silva SJ, Sankar A, Exposito F, Cai WL, Melnick MA, Robles-Oteiza C, Hinkley MM, Tsai JA, Hartley AV, Wei J, Wurtz A, Li F, Toki MI, Rimm DL, Homer R, Wilen CB, Xiao AZ, Qi J, Yan Q, Nguyen DX, J?nne PA, Kadoch C, Politi KA. Mammalian SWI/SNF chromatin remodeling complexes promote tyrosine kinase inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell. 2023 Aug 14;41(8):1516-1534.e9. doi: 10.1016/j.ccell.2023.07.005. Epub 2023 Aug 3. PMID: 37541244.