






METTL1在前列腺癌中通過tRNA衍生片段生物發生促進腫瘤發生
前列腺癌(PCa)是世界范圍內診斷頻率第二高的癌癥,也是男性癌癥相關死亡的第二大原因。RNA修飾普遍存在于轉移RNA (tRNAs)中。目前越來越多的證據表明,tRNA及其修飾酶的失調也參與了腫瘤的發生。作者的研究揭示了tRNA m7g甲基組在PCa中的復雜作用,并強調了靶向METTL1作為治療PCa的新治療策略的潛力。本文于2023年7月發布在《Molecular Cancer》,IF=37.3。
技術路線

主要研究結果
1、METTL1在人和小鼠PCa中升高
為了研究RNA修飾在PCa腫瘤發生中的潛在作用,作者采用了一種確保選擇與PCa腫瘤發生相關的RNA修飾的方法。作者專注于在五項PCa研究的數據集中鑒定132組帶注釋的RNAmodifying proteins (RMPs)的表達變化(圖1A)。此外,作者使用基因工程PCa小鼠模型將作者的分析擴展到小鼠PCa(圖1A)。作者發現原發性和轉移性PCa中表達差異最大的基因是METTL1(圖1B)。從原發性到轉移性腫瘤,METTL1的表達持續增加(圖1C),在劍橋、斯德哥爾摩和泰勒隊列中,METTL1的高表達顯示出更差的預后(圖1C)。作者還發現WDR4的表達升高(圖1C), WDR4是m7g RNA甲基轉移酶復合物的調控亞基,在其他癌癥中也過表達;然而,作者沒有發現WDR4過表達是不良預后的危險因素(圖1D)。來自當地隊列(Basurto醫院)的PCa樣本證實了METTL1和WDR4蛋白表達的增加(圖1E)。基于前列腺癌腫瘤的激素依賴性,作者進行了METTL1、WDR4和AR的表達分析,但METTL1、WDR4與AR的表達沒有明顯的相關性(圖1E)。為了證實METTL1和WDR4的表達是否與晚期腫瘤狀態相關,作者通過S6K的磷酸化狀態來測量PI3K通路的活性,在大約70%的晚期PCa患者中,S6K的磷酸化狀態發生了改變。作者發現METTL1和WDR4表達與PI3K通路激活增強呈正相關(圖1E),表明METTL1和WDR4表達在晚期PCa腫瘤中升高。綜上所述,作者的研究結果表明,METTL1是PCa中發生改變的主要表轉錄組調控因子,其過表達與預后不良相關。

圖1. METTL1在前列腺癌中高表達
2、METTL1的表達受AKT - mTOR下游信號通路調控
接下來,作者試圖闡明前列腺癌中METTL1表達上調的機制。由于雄激素受體(AR)活性增加是PCa的主要驅動因素之一,作者分析了通過受體和下游靶基因(如KLK3)的表達來測量的AR表達或活性增加是否與PCa中METTL1的表達相關。在Grasso數據集中,作者觀察到METTL1與AR和KLK3之間幾乎顯著的直接相關。在Taylor數據集中,作者發現了METTL1和AR之間的直接相關性,支持了這兩個因素之間潛在關系的概念。作者使用TGCA數據集的分析揭示了METTL1和KLK3之間的直接相關性。然而,有趣的是,在這個特定的數據集中,作者沒有發現METTL1和AR之間的顯著相關性。由于作者發現與良性前列腺增生標本相比,前列腺癌標本中METTL1蛋白表達與磷酸化- s6k呈正相關(圖1E),作者接下來分析了METTL1表達是否與PTEN表達相關,PTEN是PI3K/AKT/mTOR通路的負調節因子,在大約70%的晚期前列腺癌患者中缺失。分析的所有數據集均顯示METTL1與PTEN表達呈顯著負相關(圖2A),表明PI3K-mTOR軸調控了PCa中METTL1的表達。使用PI3K (BKM-120抑制劑)、AKT (MK2206)、mTORC1(rapamycin)和mTORC1/2 (Torin)的小分子抑制劑進一步解剖PI3K - mTORC1/2通路發現,AKT抑制降低了METTL1 mRNA的水平,mTOR抑制劑持續降低了METTL1 mRNA和蛋白的表達(圖2B-C),表明METTL1的表達是通過mTOR信號傳導調節的。接下來,作者研究了METTL1表達水平是否有助于確定PTEN缺失相關的患者生存率降低,這與臨床相關,并在臨床局部腫瘤患者中區分良性和侵襲性疾病。有趣的是,作者發現高METTL1表達突出了PTEN -低患者預后不良的子集(圖2D)。為了確定前列腺癌中METTL1的上調是否是PTEN表達低或缺失的直接后果,作者分析了野生型(WT)小鼠和Probasine-Cre x PTENflox/flox小鼠(以下簡稱PTEN - ko)前列腺組織中METTL1 mRNA和蛋白水平,這些小鼠在前列腺上皮中有條件地缺失了PTEN。這些小鼠在12周后發展為高級別腫瘤前病變,在5個月大后發展為浸潤性腺癌。作者觀察到PTEN缺失后METTL1的表達逐漸增加(圖2E-G)。有趣的是,來自小鼠前列腺腫瘤的tRNA在組織中提取的tRNA以及尿液中顯示m7g的沉積顯著增加(圖2H-K)。免疫組織化學分析進一步顯示,METTL1在小鼠前列腺癌中的表達更高,在腔細胞中也有高表達,腔細胞是最常見的上皮細胞類型,被普遍認為是人類前列腺癌的優選起源細胞(圖2L)。綜上所述,這些數據表明METTL1是mTORC1通路的下游效應物,其激活可誘導METTL1在PCa中的表達增加。

圖2. PI3K-AKT-mTORC通路介導前列腺癌中METTL1表達上調
3、METTL1優先甲基化tRNA
為了了解METTL1在PCa腫瘤發生中的作用,作者通過結合兩種轉錄組的方法確定了METTL1 RNA底物。為了鑒定PCa細胞中METTL1特異性RNA靶點,作者使用了PAR-CLIP,將光反應性核糖核苷類似物納入新生RNA中,通過紫外線交聯誘導蛋白質和RNA之間的共價鍵,然后進行下一代測序。以空載體感染多西環素誘導的細胞為對照。作者發現,與對照樣本結合的RNA相比,tRNA代表了METTL1上最豐富的RNA物種,每個基因的reads密度最高,平均中位數密度為Log2 12 rpm。(圖3)。與對照樣品相比,作者沒有觀察到HA-METTL1樣品上結合的其他RNA物種的富集(圖3A)。為了進一步驗證這些結果,作者下載了Bao等人生成的數據,分析了HEK293T細胞中METTL1結合的RNA。tRNA也是HEK293T細胞中與METTL1結合最豐富的RNA種類,平均中位密度為log28 rpm。接下來,為了精確定位tRNA中的m7g,作者使用CRISPR/Cas9敲除PCa細胞系PC3中的METTL1(圖3B)。使用抗m7g抗體的North-dot blot分析和質譜分析證實了METTL1 KO細胞tRNA中m7g的缺失(圖3B-C)。然后,作者進行了AlkAnaline-seq,以單核苷酸分辨率繪制m7g的發生圖譜,并使用METTL1 KO tRNA作為對照。高通量測序數據的分析證實了先前報道的鳥苷46可變環中強大的甲基化(圖3D, F)。作者鑒定出大約50%的同型受體為METTL1底物(圖3E, G)。綜上所述,作者的分析證實了METTL1優先甲基化PCa細胞中可變環上的tRNA。
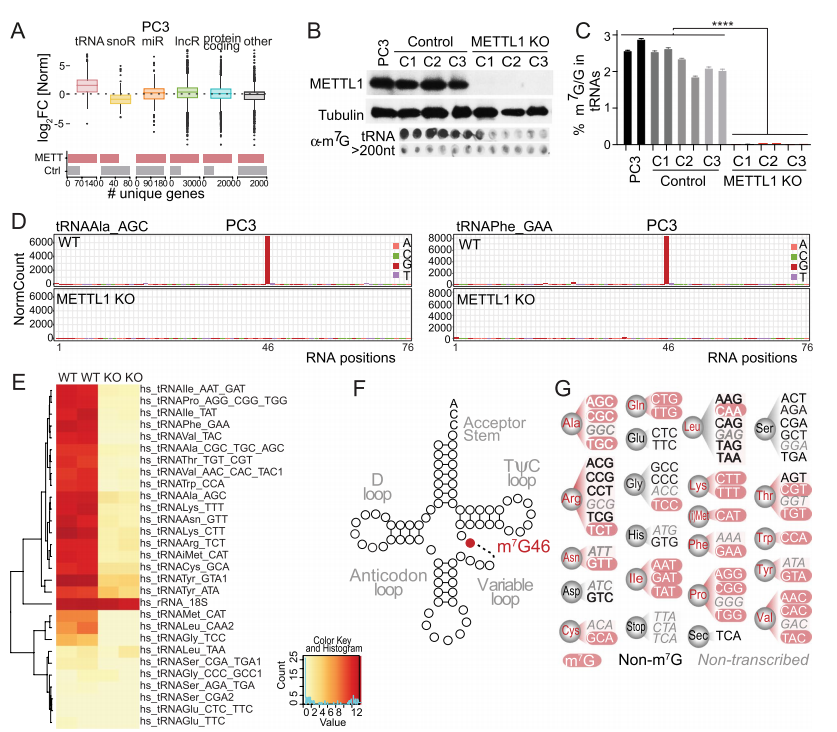
圖3. METTL1優先甲基化trna
4、METTL1介導的甲基化保護tRNA不被切割成小的非編碼RNA
在酵母和人類中,當m7g甲基化降低時,tRNA的穩定性降低。為了確定METTL1缺失可能會擾亂PCa細胞中單個METTL1靶向tRNA的水平,作者對從PC3 WT和METTL1 KO細胞中分離的tRNA進行了高通量測序。與之前的研究結果相反,作者沒有發現證據表明METTL1特異性甲基化的缺失會降低特異性成熟tRNA同受體的豐度(圖4A)。最近的報道表明,tRNA修飾保護或誘導tRNA切割成抑制性小ncRNA。作者分析了來自PC3 WT和METTL1 KO細胞的小RNA測序數據,以尋找tRNA片段的主要類別的差異。有趣的是,作者在PC3 METTL1 KO細胞中檢測到一致的5’tRNA片段富集(圖4B);然而,與WT細胞相比,大多數METTL1 KO細胞的富集程度適度增加(圖4C)。作者的分析顯示,與WT細胞相比,在所有5'tRNA片段中,一個特定的類別,即5 '末端低鳥嘌呤tRNA片段(5'TOGs),在KO細胞中顯著過度代表(log2 FC>2, p值<0.05)(圖4B;圖4C)。5 ' togs長約20或30個核苷酸,主要來源于METTL1靶tRNA Cys(產生含有5 ' togs的5個末端鳥嘌呤(5G))和Ala(產生含有5 ' togs的4G)的裂解(圖4DE)。Northern blotting證實,與WT細胞相比,PC3 METTL1 KO細胞的兩個獨立克隆中cys衍生的5 ' trf的積累較弱,但明顯較高(圖4F)。在PC3、DU145和22Rv1細胞中,METTL1下調時觀察到5'tRFs,表明5'tRFs的形成與PTEN、p53和AR狀態無關(圖4F)。一小部分tRNA切割成tRNA衍生的ncRNA是對應激的保守反應,tRNA修飾保護它們免受應激誘導的切割。在應激反應中,tRNA切割在METTL1 KO細胞中比在WT細胞中更為突出,早在氧化應激暴露2小時達到峰值,在應激刺激8小時后下降,可能是因為無法解決應激導致細胞死亡增加(圖4G, H)。總之,作者的數據表明,METTL1介導的甲基化是一種保守的機制,它調節了PCa細胞在應激反應中源自5’tRNA片段的一類新型小ncRNA的生物發生,而不管它們的遺傳狀態如何。
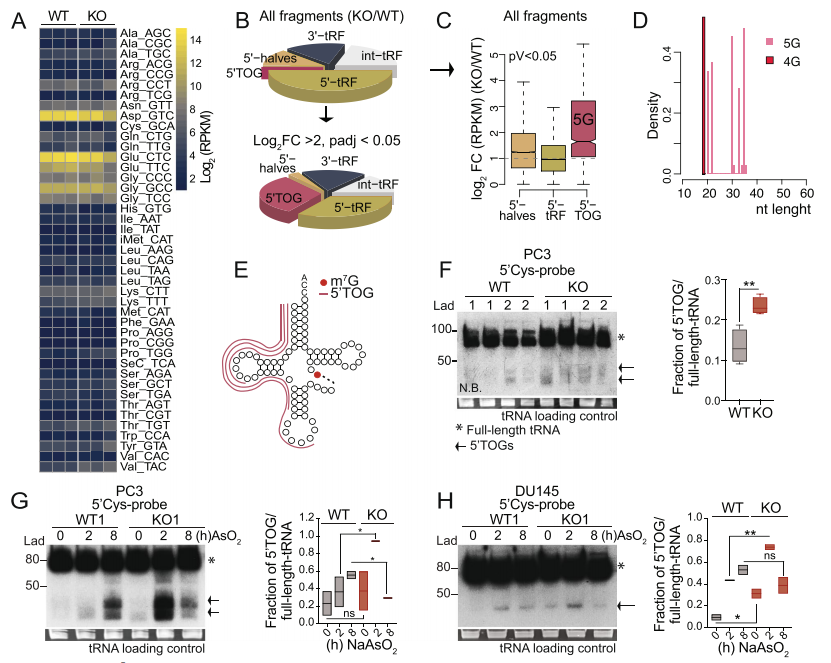
圖4. tRNA中缺乏m7g甲基化導致5'tRNA片段積累
5、METTL1的缺失通過tRNA片段生物發生抑制翻譯起始
由于已知5'TOGs抑制全局翻譯,作者接下來研究了5'TOGs的積累是否導致了PC3 METTL1缺失細胞的翻譯改變。作者證實,與WT細胞相比,METTL1 KO細胞中通過o -丙基嘌呤霉素(OP-puro)摻入測量的蛋白質翻譯減少(圖5A)。細胞應激反應中5’tRNA片段的產生在應激反應機制中起關鍵作用。這個過程有助于抑制整體蛋白質合成,使細胞能夠恢復并在壓力條件下生存。為了確定METTL1的去除是否會影響應激反應,作者測量了氧化應激誘導后的蛋白質合成速率。m7g的缺失顯著抑制了蛋白合成,并且這一過程與eIF2α磷酸化狀態無關。接下來,作者試圖確定METTL1 KO細胞獨特的翻譯抑制及其對5'TOGs的依賴的分子基礎。先前的證據表明,5'TOGs通過取代mRNA帽上的翻譯起始復合物eIF4A/G/E和調控因子YB1和PABP1的組分,從而損害翻譯起始。為了確定翻譯起始因子是否從PC3 METTL1 KO細胞中7-甲基-鳥苷化(m7g)覆蓋的mRNA中轉移,作者分析了翻譯起始因子對m7g -帽狀結構被的sepharose beads的親和力。作者發現,與WT細胞相比,METTL1 KO細胞中eIF4G和PABP1的m7 g -cap親和力顯著降低(圖5B)。這表明,5'TOGs積累的增加破壞了翻譯起始復合物某些因子的組裝和結合的穩定性,導致METTL1 KO細胞中的翻譯受到抑制。作者進一步評估了在WT細胞中表達的合成5'TOG是否能夠結合翻譯起始因子和調節因子,以及它們對這些因子的親和力是否可以通過合成的逆補體5'TOG RNA(或抗tog)來阻斷。為此,作者用5 '生物素化的5'TOG RNA(包含PC3 METTL1 KO細胞中最豐富的5'TOG序列)轉染PC3 WT細胞,并添加或不添加抗tog。在去除5 '生物素化-5 ' togs后,作者發現在抗togs存在下,PABP1和YB1對5'TOG的親和力顯著降低(圖5C)。接下來,作者研究了人工合成的5'TOGs是否可以在WT細胞中表型化翻譯起始復合物的位移,以及抗tog RNA是否可以在METTL1 KO細胞中挽救這種觀察到的效應。作者發現,在轉染5'TOGS的WT細胞中,PAPB1與m7g -cap的結合被取代,但在轉染抗tog RNA的METTL1 KO細胞中,PAPB1與m7g -cap的親和力顯著增加(圖5D)。總的來說,這表明5'TOGs的形成通過取代PAPB1來抑制蛋白質翻譯。有趣的是,PAPB1先前已被證明對其他細胞類型的5'TOGs具有很強的親和力。因此,作者的研究結果證實,METTL1 KO細胞中表達的5'TOGs可以取代mRNA帽上的翻譯調節因子,并為METTL1介導的翻譯抑制的分子基礎提供了關鍵見解。

圖5. METTL1下調抑制體內蛋白合成、增殖和腫瘤生長
6、METTL1下調可抑制前列腺腫瘤在體內和體外的生長
鑒于METTL1 KO細胞合成的蛋白質較少,作者假設METTL1抑制可以降低細胞和腫瘤的生長。事實上,作者觀察到METTL1的敲低會損害PC3、DU145和22Rv1細胞的生長(圖5E)。METTL1下調還會減少細胞分裂,誘導細胞周期阻滯,增加細胞凋亡,損害球體形成能力,并顯著降低腫瘤異種移植物的生長和增殖(圖5F-I)。因此,作者的數據表明,下調METTL1可以有效地抑制腫瘤生長,有力地支持METTL1在調節PCa進展中起關鍵作用。接下來,作者研究了METTL1甲基化酶活性是否足以促進細胞生長。作者在PC3 METTL1 KO細胞中重新表達了野生型METTL1 (WT)和催化死亡突變體(AFPA)(圖5J-L)。與用空載體(eV)轉導的KO細胞相比,重新表達WT METTL1的KO細胞的增殖和球體形成能力增強。相比之下,無催化活性突變體的表達未能促進METTL1 KO細胞生長和球體形成能力(圖5M-N)。由于METTL1的催化活性得到了與調控亞基WDR4形成復合物的支持,并且WDR4在PCa中過表達(圖1C, E),因此作者測試了WDR4的致癌潛力。誘導沉默WDR4并不會降低細胞或腫瘤的生長。綜上所述,這些結果表明METTL1以酶活性依賴的方式促進PCa細胞的生長,但不依賴于WDR4。為了進一步確定5'TOG是否足以誘導生長停滯和凋亡,作者用5'TOG轉染PC3 WT細胞,用抗tog轉染METTL1 KO細胞,并測量細胞凋亡和增殖。作者檢測到WT細胞轉染5'TOG RNA后,凋亡顯著增加,增殖顯著減少,而METTL1 KO細胞轉染抗togs后,凋亡顯著減少,但不顯著,增殖顯著增加(圖50,P)。總之,作者的數據表明,METTL1失活和5'TOGs是誘導生長停滯的必要條件。METTL1催化活性版本和抗togs的重新表達可以部分修復METTL1 KO細胞中觀察到的缺陷。
7、METTL1丟失激活IFN信號通路
鑒于翻譯受到METTL1抑制的影響,作者探討了METTL1缺失對翻譯體即刻變化的影響。為此,作者利用OP-puro特性來標記新生蛋白,隨后將其偶聯到生物素包被的微球上,然后進行微球上的消化和LC-MS /MS(圖6A)。基因本體(GO)術語富集分析發現,與細胞分裂和有絲分裂相關的基因翻譯減少,證實了觀察到的METTL1 KO細胞增殖缺陷(圖6B)。出乎意料的是,作者還發現METTL1 KO細胞中與應激反應相關的轉錄本翻譯增加,包括I型和II型干擾素(IFN)信號通路、免疫效應過程和分解代謝過程(圖6B)。翻譯的差異反映在PC3 METTL1 KO細胞的整體蛋白質組組成中,但與RNA表達水平無關,這表明轉錄后或翻譯調控是導致METTL1 KO細胞中不相關的RNA -蛋白表達水平的原因(圖6C)。為了進一步測試METTL1 KO細胞中某些轉錄本的翻譯效率是否存在差異,作者進行了多體分析。這種方法使作者能夠確認METTL1 KO細胞中活性多體的形成減少,同時整體蛋白合成減少(圖6D)。對METTL1 KO細胞多體部分mRNA富集的分析顯示,與干擾素信號相關的特定轉錄物的翻譯增加,如干擾素調節因子9 (IRF9)和干擾素刺激基因15 (ISG15)(圖6D)。作為翻譯變化特異性的對照,作者沒有發現在METTL1 KO細胞的多體部分中富集GAPDH、Catenin β、KIF20A或STAT1等轉錄本(圖6D)。綜上所述,作者的研究結果表明,METTL1介導的tRNA甲基化引導著一個獨特的翻譯程序。由于5'TOGs可以重新編程翻譯機制,以支持癌細胞所需的翻譯程序,作者接下來研究了METTL1 KO細胞的翻譯變化是否由5'TOGs的生物發生增加介導。為此,作者用5'TOG和抗tog RNA轉導PC3 WT和METTL1 KO細胞,并評估蛋白質表達的變化。作者發現,WT細胞中轉染5'TOGs增加了IRF9和ISG15蛋白的表達,而在METTL1 KO細胞中轉染抗tog RNA輕微但顯著地降低了這兩種蛋白的表達(圖6E)。作者還觀察到METTL1 KO細胞和異種移植物中STAT1的蛋白表達和磷酸化增加(圖6E),但其表達的增加與翻譯的增加無關(圖6D),而是轉錄依賴的,這表明STAT1表達的增加是由METTL1 KO細胞中IFN信號通路的激活誘導的。考慮到STAT1激活的抗增殖和促凋亡作用,作者測試了下調STAT1是否可以逆轉METTL1 KO PC3細胞的生長缺陷。作者發現,在STAT1敲除后,PC3 METTL1 KO細胞的凋亡減少,增殖增加(圖6F, G),這表明METTL1抑制的抗增殖作用可能部分是通過激活STAT1信號通路介導的。綜上所述,作者的數據表明,METTL1介導的tRNA甲基化和tRNA片段生物發生誘導了激活IFN信號通路的翻譯程序。為了證實METTL1的高表達與人類PCa樣本中IFN通路活性的降低相關,作者在三個PCa表達數據集中評估了ISGs的表達。綜上所述,作者的數據表明,在PCa細胞和腫瘤中,METTL1的表達與IFN通路的激活呈負相關,METTL1的抑制可翻譯激活IFN信號通路。
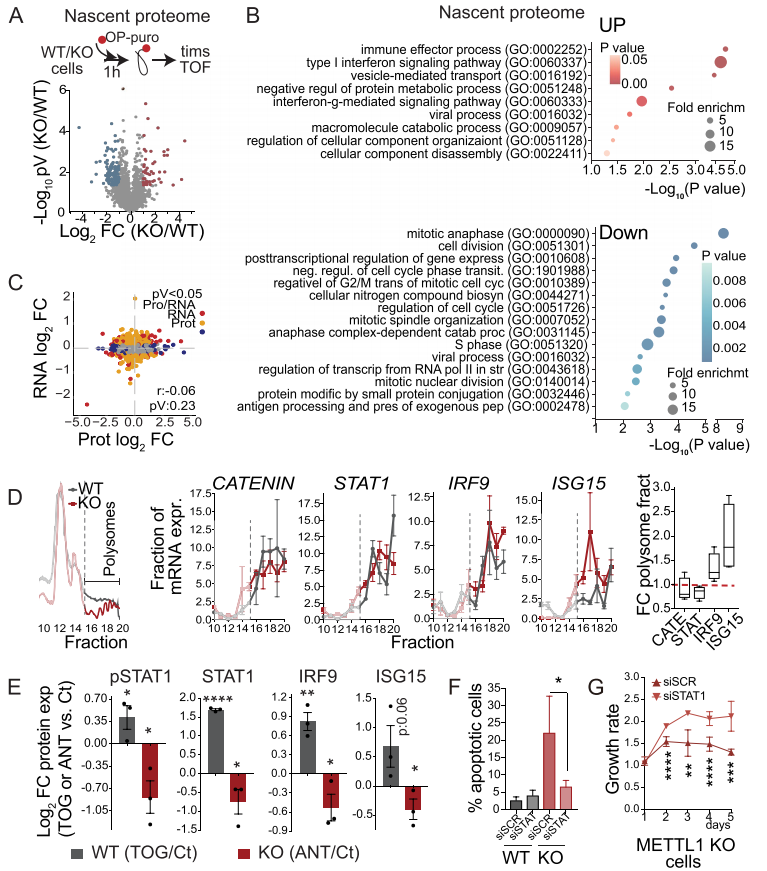
圖6. m7G tRNA甲基化的缺失導致不同的翻譯程序
8、前列腺癌中METTL1的低表達與促炎免疫細胞極化增加相關
最近,表觀遺傳靶向治療已被證明在幾種癌癥類型中觸發IFN抗病毒反應,包括PCa,引發先天免疫反應并導致許多細胞因子的產生。為了闡明METTL1抑制是否可以通過激活IFN信號通路引發先天免疫反應,作者研究了METTL1下調是否會改變PCa細胞中細胞因子的表達和分泌。總體而言,在METTL1 KO細胞中,細胞因子組成分析顯示,參與促炎活性的細胞因子分泌增加,并使巨噬細胞極化為m1樣內型,包括粒細胞-巨噬細胞集落刺激因子(GM-CSF)和腫瘤壞死因子α (TNF-α)(圖7A)。METTL1的去除也誘導了抗炎細胞因子的下調,包括巨噬細胞或集落刺激因子(M-CSF)、IL10和IL13(圖7A),可使巨噬細胞極化為m2樣內型。這些數據表明,腫瘤細胞中METTL1的抑制可以使腫瘤微環境(TME)中的免疫細胞向細胞毒性殺瘤內型分化。分子技術的出現,如單細胞RNA測序(scRNA-seq)和空間轉錄組學,使審計腫瘤免疫微環境的組成成為可能。無監督的scRNAseq聚類和空間轉錄組學分析顯示,在先天免疫群體中,巨噬細胞在PCa中所占比例最大。腫瘤相關巨噬細胞(tam)通常表現出m2樣或促腫瘤巨噬細胞的表達模式特征,是支持腫瘤發生的TME的主要組成部分。眾所周知,它們促進癌細胞增殖、侵襲性、血管生成和免疫耐受,同時也有助于對標準治療、免疫治療和化療的耐藥性。因此,迫切需要開發能夠消耗巨噬細胞或誘導其向m1樣或抗腫瘤狀態再極化的臨床藥物。考慮到這些情況,以及觀察到表達METTL1的細胞分泌的細胞因子已知可驅動巨噬細胞向m2樣內型極化,作者重點闡明了METTL1在巨噬細胞極化中的作用。作者用PC3 WT或METTL1 KO細胞的條件培養基(cm)培養人單核細胞系THP1,并研究了m1樣和m2樣巨噬細胞內源性標記物的表達(圖7B)。tSNE分析顯示,WT細胞中的c.m.誘導巨噬細胞向m1樣內型轉變,而抑制PCa細胞中的METTL1則誘導巨噬細胞向m1樣內型轉變(圖7B)。此外,PC3 METTL1 KO細胞cm存在下培養的外周血巨噬細胞對CD3+T細胞的增殖和遷移具有更高的誘導作用(圖7C, D)。總之,作者的數據表明,METTL1在癌細胞中的抑制可能會刺激TME的細胞毒性和抗腫瘤炎癥反應。為了在體內驗證作者的發現,作者使用免疫組織化學分析評估了人類前列腺腫瘤的腫瘤內免疫細胞組成。分析表明,在PCa石蠟樣品中,m2樣巨噬細胞浸潤與METTL1表達之間存在顯著的直接相關,但m1樣巨噬細胞浸潤呈相反趨勢。同樣,CD8+T細胞浸潤與METTL1表達呈負相關(圖7E)。總之,作者的研究結果表明,抑制前列腺癌細胞中METTL1的表達可以在體外引發細胞毒性免疫反應,并進一步支持人前列腺癌中METTL1的低表達與腫瘤內細胞毒性免疫細胞的增加有關。

圖7. METTL1在PCa中的低表達與細胞毒性浸潤增加和對ICB治療的良好反應相關
9、METTL1抑制增強了前列腺癌患者對免疫檢查點阻斷治療的應答
PTEN-KO小鼠前列腺上皮中METTL1雜合缺失(PTEN-KO/METTL1+/-)和METTL1條件缺失(PTEN-KO/ mett1flox /flox)導致腹側和前葉腫瘤體積顯著減少,PTEN-KO/METTL1+/+小鼠的腫瘤體積始終較大(圖7F)。基于其特征是誘導m1樣巨噬細胞極化,以及體外CD8+T細胞的增殖和遷移增強(圖7AD),作者隨后的研究重點是分析METTL1缺陷前列腺腫瘤中巨噬細胞和CD8+T細胞的組成。免疫組織化學分析顯示,與METTL1+/+腫瘤相比,m1樣巨噬細胞(iNOS+)的瘤內浸潤顯著增加,而m2樣巨噬細胞(Arg1/CD68+)的浸潤則顯著增加,并且METTL1+/-和METTL1+/+和METTL1 /flox腫瘤中CD8+T細胞增加(圖7G)。細胞毒性T細胞,以其CD8的表達而聞名,具有直接識別和消除癌細胞的非凡能力,使其成為抗癌免疫反應中最有效的效應器。為了確定小鼠METTL1缺失的抗腫瘤作用是否由于腫瘤內細胞毒性CD8+T細胞浸潤增加,作者檢測了抗CD8α抗體處理PTEN-KO/METTL1+/+和PTEN-KO/METTL1fl/fl小鼠全身消除CD8+T細胞的效果。為了闡明METTL1抑制對免疫腫瘤組成重編程的影響,作者分析了免疫調節分子,包括細胞因子和趨化因子,作為腫瘤中存在的免疫細胞的替代標記物。作者的研究結果揭示了細胞因子組成的顯著變化,其特征是促腫瘤細胞因子的分泌減少。此外,作者觀察到在PTEN-KO/ mePCa1fl /fl腫瘤中,已知與免疫檢查點阻斷(ICB)治療有利應答相關的細胞因子分泌增加(圖7H)。這些發現表明,抑制METTL1可能有助于改善ICB治療結果的免疫微環境,突出了靶向METTL1作為增強抗腫瘤免疫反應的治療策略的潛力。為了確定mett1缺陷腫瘤的腫瘤內細胞因子組成是否可以增強ICB治療的療效,作者用抗pd1和抗ctl4a抗體治療小鼠。與未治療的小鼠相比,PTEN-KO/METTL1+/+小鼠在ICB治療后未顯示腫瘤體積減少,而PTEN-KO/ mett1flox /flox小鼠在ICB治療后腫瘤體積顯著減少(圖7I)。作者還研究了METTL1表達水平是否可以預測患者ICB治療的療效。使用ROC繪圖儀平臺,作者分析了METTL1在抗pd1治療的幾種腫瘤類型中的表達水平。作者發現,在乳腺癌、結直腸癌、卵巢癌和膠質母細胞瘤患者中,對ICB治療有反應的METTL1表達低于無反應的METTL1表達,這表明高METTL1表達預示著對ICB治療的不良反應(圖7J)。總之,作者的研究結果表明,METTL1的表達水平可以決定腫瘤內細胞毒性免疫浸潤和ICB治療的成功。
結論
作者的研究為METTL1在前列腺癌(PCa)中的致癌功能和過表達提供了令人信服的證據。作者還通過tRNA片段生物發生發現了一個新的表達調控和選擇性翻譯控制層。通過抑制METTL1介導的甲基化,作者能夠增加一類新的ncRNA (5'TOGs)的生物發生,這些ncRNA調節癌細胞中干擾素信號通路的激活。此外,癌細胞中METTL1的抑制導致細胞毒性免疫細胞的浸潤增加,從而將免疫抑制的前列腺TME轉變為殺瘤內型。這些結果表明,單獨靶向METTL1或聯合免疫檢查點抑制劑具有開發有效治療策略的巨大潛力,特別是對于治療選擇有限的去勢抵抗性前列腺癌患者。
實驗方法
細胞培養、PI3K通路抑制和DHT治療、患者樣本收集、人類前列腺腫瘤數據集的計算表達和突變負荷分析、穩定細胞系的克隆和生成、WB、RNA提取、qPCR、斑點印跡、免疫組化、免疫熒光、PAR?CLIP、AlkAniline?seq、tRNA?seq、Northern blotting、流式細胞術、Cap binding assay、轉染、pulldown、增殖實驗、外周血T細胞分離、增殖和遷移試驗
參考文獻
García-Vílchez R, A?azco-Guenkova AM, Dietmann S, López J, Morón-Calvente V, D'Ambrosi S, Nombela P, Zamacola K, Mendizabal I, García-Longarte S, Zabala-Letona A, Astobiza I, Fernández S, Paniagua A, Miguel-López B, Marchand V, Alonso-López D, Merkel A, García-Tu?ón I, Ugalde-Olano A, Loizaga-Iriarte A, Lacasa-Viscasillas I, Unda M, Azkargorta M, Elortza F, Bárcena L, Gonzalez-Lopez M, Aransay AM, Di Domenico T, Sánchez-Martín MA, De Las Rivas J, Guil S, Motorin Y, Helm M, Pandolfi PP, Carracedo A, Blanco S. METTL1 promotes tumorigenesis through tRNA-derived fragment biogenesis in prostate cancer. Mol Cancer. 2023 Jul 29;22(1):119. doi: 10.1186/s12943-023-01809-8IF: 37.3 Q1 . PMID: 37516825IF: 37.3 Q1 ; PMCID: PMC10386714IF: 37.3 Q1 .