






沒跑了,TP53?R249S突變增加患癌風險——人肝臟類器官權威驗證
如今,肝細胞癌(HCC)的主要基因組驅動因素已得到廣泛認可,但建立它們在人類HCC發生過程中的作用模型仍然很少。本文作者利用人肝臟類器官作為實驗系統,從TP53缺失和L3環R249S突變的遺傳病變中模擬人類肝癌發生的早期階段。此外,HCC細胞系的染色質免疫沉淀測序(ChIP-seq)揭示了TP53缺失導致的抑瘤功能喪失和p53突變導致的功能增益活動引發HCC的重要功能。本文于2023年9月發表在《Hepatology》IF:13.5期刊上。
技術路線

主要實驗結果
1、ChIP-seq揭示L3突變體在染色質重塑和應激反應中的功能獲得作用
在HCC中,TP53經常因錯義突變而發生改變,占突變病例的一半以上(圖1A),且大多集中在DNA結合域(DBD)Loops 1-3 內(圖1B)。在TCGA數據集中所有攜帶TP53突變的HCC中,約60%同時攜帶TP53缺失和一個額外的改變蛋白質的錯義突變(TP53missense/-)(圖1C)。為系統評估HCC中TP53錯義突變對轉錄激活的潛在增益,通過ChIP-seq實驗,對四個TP53突變體(包括兩個L3環突變(TP53R249S/-和TP53S241F/-)和兩個非L3環突變(TP53L145R/-和TP53H168R/-)的TP53結合和賴氨酸H3K27ac的全基因組模式進行了數據分析。結果發現,無論TP53突變位點如何,WT TP53的全基因組結合在這4種突變體中大部分都喪失了,這意味著TP53錯義突變的腫瘤中普遍喪失了傳統的TP53轉錄活性(圖1D)。值得注意的是,與WT TP53和其他兩個非L3環突變體相比,一組TP53 結合峰僅在L3環突變體中被發現(圖1E)。此外,還觀察到R249S突變體特有的啟動子近端TP53結合峰,這提供了其他轉錄調控特性的可能性(圖1E)。相反,在非L3環突變體中觀察到了罕見的獨特結合峰。富集的H3K27ac信號在 L3環突變體結合區域的共定位進一步表明這些峰與活躍的轉錄有關(圖1E)。此外,為說明潛在的轉錄因子,作者在內源性TP53R249S/-和TP53S241F/-細胞中定義了紅細胞轉化特異性和堿性螺旋-環-螺旋家族的核心結合基序(圖1F)。
為確定這些TP53突變體的功能,使用靠近這三組TP53結合峰的基因進行通路富集分析。如預期的,WT-TP53主要參與細胞凋亡、細胞周期和DNA損傷的調節(圖1Gi),這支持TP53在HCC中的抑瘤作用。L3環突變體與染色質重塑和生物合成過程相關的基因結合(圖1Gii),而在TP53R249S/-中發現的獨特結合峰則富集于其他通路,包括應激反應和蛋白質修飾(圖1Giii)。
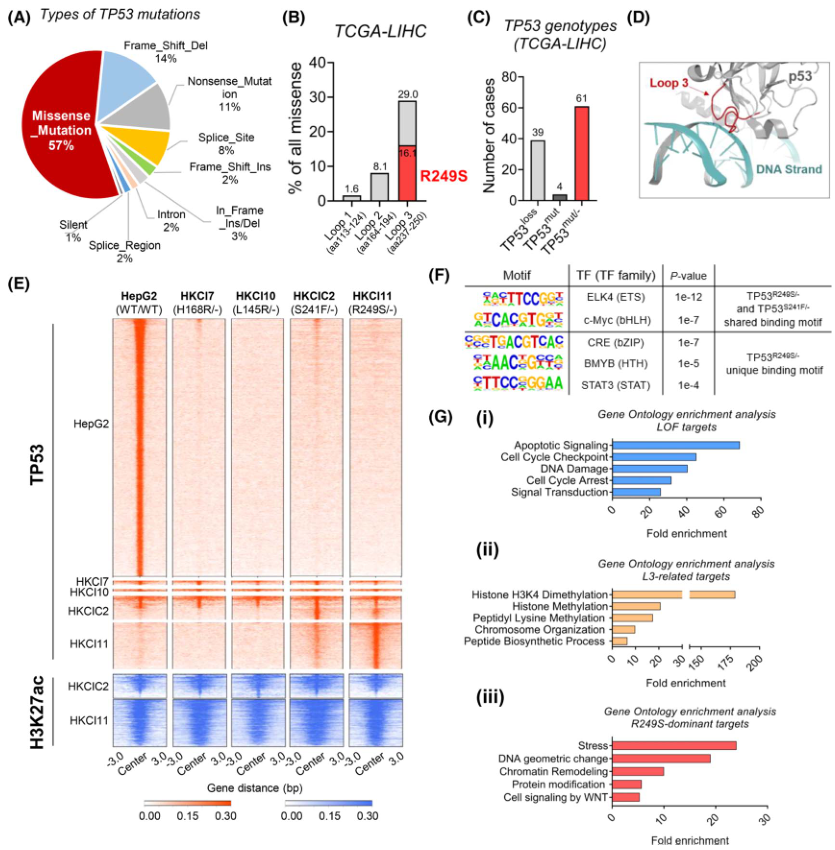
圖1 TP53基因型的ChIP測序
為證實ChIP-seq發現的L3突變體新的結合位點,在HepG2(TP53WT/WT)、HKCIC2(TP53S241F/-)和HKCI11(TP53R249S/-)細胞中進行染色質免疫沉淀-qPCR,以驗證TP53突變體與富集靶標的轉錄起始位點(TSS)區域的直接結合。首先,為確認TP53 L3突變體的功能缺失特性,測試了與內在凋亡信號轉導相關的靶標EDA2R、PHLDA3和CDKN1A。TP53R249S/-和TP53S241F/-似乎不與這些基因結合,而HepG2 TP53WT/WT則顯示出與TSS區域的富集結合(圖2Ai、Aii)。接下來,在TP53R249S/-突變體和TP53S241F/-突變體中發現,參與染色體組織調控的L3相關靶標RCOR3和UBE2I顯示出很強的TSS占有率,而在TP53WT/WT突變體中則沒有發現(圖2B)。此外,R249S顯性靶標的功能還包括應激反應、CLTC和PSMF1、紡錘體組織SUN2以及染色質重塑BPTF在TP53R249S/-與TSS區的結合均顯示出顯著的富集,與TP53S241F/-和TP53WT/WT 相比(圖2C)。
為證實L3突變體增強了基因表達,作者在配對的HCC和鄰近非腫瘤肝臟的獨立原發性隊列中進行驗證。這些標本根據目標捕獲測序或Sanger 測序驗證的TP53狀態進行分組,包括WT(TP53WT/WT)、無效(TP53-/-)、TP53R249S、L3 環內的TP53錯義突變(其他L3)和L3環外的錯義突變(non-L3)。除TP53WT/WT 外,所有組的功能缺失靶點都明顯減少(圖2D)。在TP53 L3環突變的標本中,L3環靶點明顯升高(圖2E)。值得注意的是,與TP53WT 和其他突變組相比,TP53R249S病例中6個R249S顯性靶標明顯升高(圖2F)。
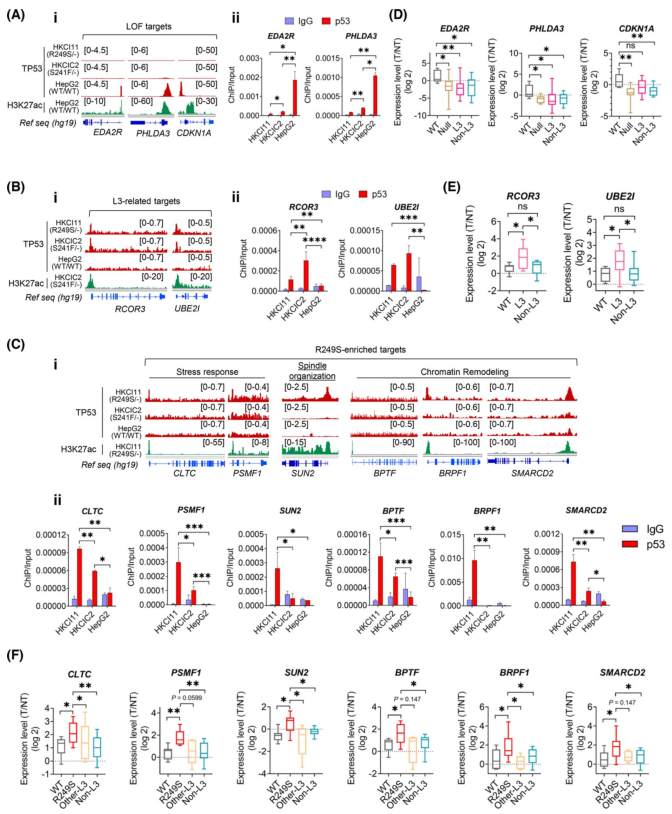
圖2 功能喪失(LOF),回路L3和R249S富集靶標的驗證
2、TP53 KO和R249S產生原發性肝類器官和致瘤誘導
為了解TP53KO和TP53R249S在HCC中的致癌作用,作者從新鮮的非腫瘤肝組織中培養了三個肝臟器官模型,即肝臟類器官模型1(Liver.Org 1)、肝臟類器官模型2(Liver.Org 2)和肝臟類器官模型3(圖3A)。為模擬TP53R249S/-基因型,首先應用CRISPR/Cas9系統的兩個single‐guided RNA在三個肝臟器官組織中 KO TP53(圖3B)。隨后通過pLV-Hygro-TP53R249S慢病毒進行相應的TP53 R249S 過表達(圖3B)。TP53蛋白的丟失和獲得被WB所證實(圖3C)。從形態上看,WT(TP53WT)肝臟類器官的明視野成像和組織學染色都保持了單層分區的一致性(圖3D)。相比之下,TP53KO和R249S過表達(TP53R249S)的類器官出現了不同的形態,可見增厚的管壁侵入管腔,并呈現多形性惡性特征,包括發育不良、色素沉著、不典型和頻繁的有絲分裂、極性喪失和核與細胞質比率增加(圖3D)。對Liver.Org 1的延時成像進一步驗證了TP53KO和TP53R249S類器官中出現的生長模式變化的異常形態特征。與TP53WT對照組有機體的導管結構(圖3E)相比,觀察到TP53蛋白缺失后形成的緊湊有機體簇(圖3E)。有趣的是,R249S過表達的類器官表現出非典型生長,類器官表面有大量芽狀突起(圖3E),這與HCC類器官的生長模式非常相似。通過高通量成像檢測,量化了不同TP53狀態下的形態差異。與組織學一致,單層有機體常見于TP53WT組,而腫瘤喜歡的多層或發育不良的有機體則廣泛見于TP53KO和TP53R249S Liver.Org 1和Liver.Org 2組(圖3F)。
此外,為評估TP53缺失或R249S突變是否會帶來生長優勢,注意到所有TP53WT對照組器官組織都在20次傳代過程中停止生長(圖3G)。令人驚訝的是,TP53KO和TP53R249S類器官在繁殖過程中繼續增殖和擴張,在超過50次的傳代過程中,每周的分裂率始終為1:4(圖3G)。據報道,干性是促進細胞壽命的因素之一,因此評估了三個肝臟類器官中肝癌干性標志物CD44和CD133的表達。與TP53WT對照組相比,TP53KO和TP53R249S類器官中CD44和CD133的表達明顯增加(圖3H),表明癌癥相關干性能力明顯受到刺激。
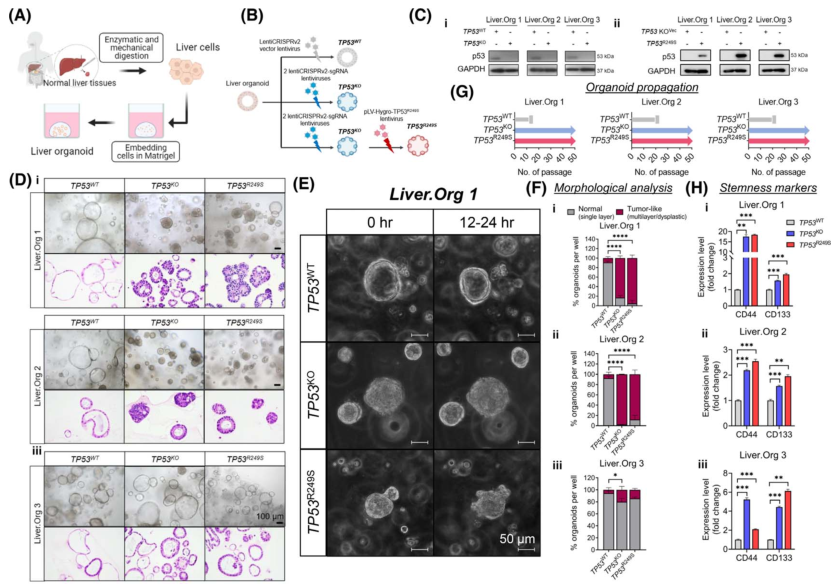
圖3 TP53基因敲除(TP53KO)和TP53R249S的肝類器官表現出癌前特征
3、模型肝臟類器官中TP53R249S引發的腫瘤
接下來探討突變體R249S和癌癥相關應激在啟動肝癌發展中的作用。首先驗證了TP53功能缺失靶基因和R249S顯性候選基因的表達,這些基因與編輯肝臟類器官中的應激反應和紡錘體組織有關。與在原發性HCC中發現的表達一致,在R249S高表達的肝臟組織細胞中檢測到R249S優勢靶基因的明顯上調(圖4A)。在分別過表達TP53 WT和R249S的Hep3B(TP53-/-)細胞中進行雙熒光素酶檢測,進一步確定了R249S突變體的直接轉錄效應。與TP53 WT對照組相比,TP53 R249S表達細胞中兩個與應激相關的R249S優勢靶標CLTC和PSMF1的啟動子活性明顯升高(圖4B)。錨定非依賴性實驗提示了一鍋可能的腫瘤發生其實作用,表現為TP53R249S類器官中有大量致密的大球體,而TP53KO類器官中有大量的球體簇,而對照類器官中只有微小的散發性球體(圖4C)。異種移植進一步證實了TP53R249S的致瘤潛力。2個月后,皮下類器官異種移植(ODXs)顯示,與TP53缺陷類器官(17%)和WT對照(0%)相比,R249S高表達肝臟類器官(37%)的病變形成率增加(圖4D)。組織學檢查顯示這些病變具有惡性特征,顯示出腺體形成的非典型細胞,這些細胞表現出核多形性高色差、偶見空泡狀胞質和突出的核小體(圖4E)。這些發現表明,p53缺失和R249S基因突變同時存在是一種惡性轉化。
接下來,研究R249S相關應激反應在腫瘤傾向中的促進作用。有幾種類型的應激,如ER應激、氧化應激和代謝應激,會積極調控癌癥的發展。使用不同的應激特異性刺激劑,包括ER應激誘導劑曲卡霉素、氧化應激誘導劑過氧化氫、依托泊苷產生的基因毒性應激以及營養缺乏培養基產生的代謝應激誘導劑。值得注意的是,TP53R249S只對曲奈霉素有明顯的耐受性,而對其他誘導劑沒有耐受性(圖4F)。鑒于ER應激與活性氧(ROS)誘導之間的相互串擾,作者評估了ER應激刺激下的ROS水平。CellROX Red信號在TP53R249S類器官組織中被模糊地檢測到,而在對照組中則被清晰地識別到(圖4G),這意味著R249S的功能獲得作用產生了ROS平衡水平。為確認ER應激耐受性與R249S顯性應激靶標PSMF1之間的聯系,通過敲除PSMF1對TP53R249S肝臟類器官進行拯救實驗(圖4H)。PSMF1沉默的肝臟類器官恢復了對妥卡霉素的自發敏感性,IC50值比對照低4-8倍(圖4I)。
由于錯誤折疊蛋白的積累是ER應激的標志,作者檢測了PSMF1下調的類器官中未折疊蛋白反應(UPR)的關鍵傳感器和介質的表達。與對照相比,PSMF1缺陷型TP53R249S類器官中ATF6、GRP94、CHOP和GADD34 GADD的表達量在使用曲卡霉素處理時明顯增加(圖4J),這意味著PSMF1在通過防止UPR激活來緩解ER應激方面具有重要能力。
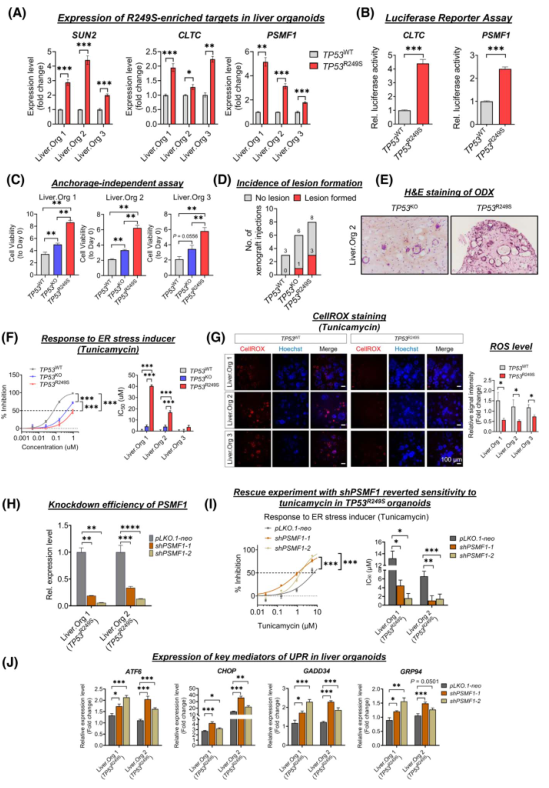
圖4 TP53R249S肝類器官中的腫瘤發生
4、通過染色質重塑靶向TP53R249S的一個治療窗
考慮到R249S顯性靶標中"染色質重塑"GO的富集(圖1Giii),作者首先確定了TP53R249S肝臟類器官中R249S顯性靶標(BPTF、BRPF1和SMARCD2)的顯著上調(圖5A)。由于硫鳥嘌呤是一種抑制DNA合成和甲基化的表觀遺傳藥物,所以作者在體外探討了硫鳥嘌呤在TP53R249S類器官中的藥理作用。結果顯示,與WT對照組相比,TP53R249S類器官更容易受到硫鳥嘌呤的影響,其敏感性在IC50值方面顯著增加4.5-7倍(圖5B)。
為進一步證實硫鳥嘌呤的體內療效,利用Liver.Org 1和Liver.Org 2的 TP53R249S 類器官建立ODX 模型。當腫瘤體積達到約50立方毫米時,隨機給小鼠腹腔注射硫鳥嘌呤(1.5 mg/kg)或藥物,連續注射14天。結果顯示,TP53R249S ODX對硫鳥嘌呤更敏感,與藥物對照組相比,治療14天后腫瘤體積明顯縮小(圖5C、D)。兩個治療組的體重沒有明顯差異(圖5E)。所有主要的器官組織學也都正常,這表明硫鳥嘌呤的副作用可以忽略不計(圖5F)。總之,研究證明表觀遺傳藥物硫鳥嘌呤在體外和體內針對與TP53R249S相關的功能獲得脆弱性的治療潛力。
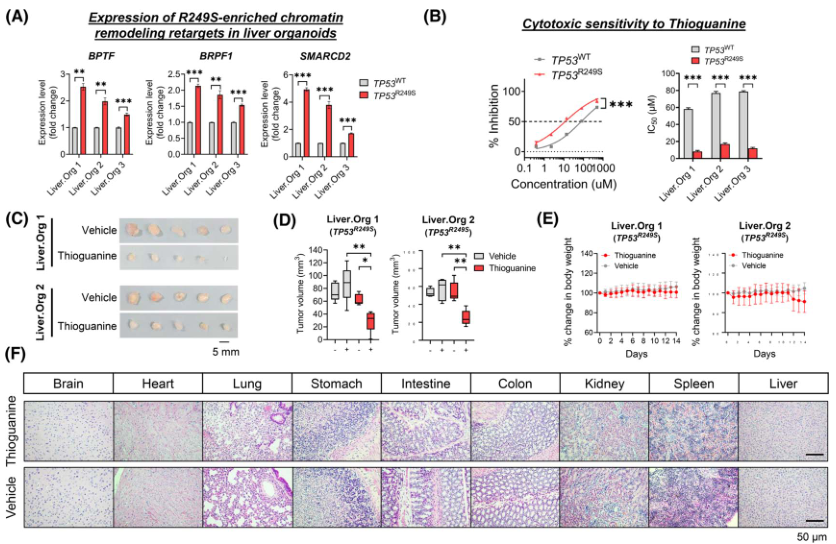
圖5 靶向TP53R249S的一個治療窗
總之,本文發現TP53缺失和L3 loop突變會產生不同的致瘤效應,它們共同賦予正常肝細胞早期克隆優勢和促生存功能(圖6)。

圖6 圖形摘要
實驗方法
HCC臨床樣本收集,肝臟類器官培養,慢病毒轉染類器官,類器官形態觀察,壓力敏感實驗,肝臟類器官來源的異種移植瘤
參考文獻
Lam YK, Yu J, Huang H, Ding X, Wong AM, Leung HH, Chan AW, Ng KK, Xu M, Wang X, Wong N. TP53 R249S mutation in hepatic organoids captures the predisposing cancer risk. Hepatology. 2023 Sep 1;78(3):727-740. doi: 10.1002/hep.32802. Epub 2022 Oct 11. PMID: 36221953