






m6A調節的腫瘤糖酵解“集合”--癌癥治療的“新星”
欄目:最新研究動態
發布時間:2023-12-22
研究者認為m6A修飾糖酵解在腫瘤治療中具有重要意義和潛力......
糖酵解重編程是癌癥最重要的特征之一,在癌癥的發展中起著不可或缺的作用。在癌細胞中,葡萄糖代謝的變化滿足了自身增殖、血管生成和淋巴管生成、轉移的需要,也影響了腫瘤的免疫逃逸、預后評價和治療效果。RNA的n6-甲基腺苷(m6A)修飾在真核細胞中廣泛存在。動態和可逆的m6A修飾廣泛參與腫瘤干細胞更新和分化、腫瘤治療抵抗、腫瘤微環境、腫瘤免疫逃逸和腫瘤代謝的調控。近年來,越來越多的證據表明m6A修飾可以通過多種方式影響腫瘤的糖酵解過程,從而調節腫瘤的生物學行為。本文綜述了糖酵解在腫瘤發生發展中的作用,并詳細闡述了m6A修飾通過調節糖酵解對不同腫瘤的深遠影響。研究者認為m6A修飾糖酵解在腫瘤治療中具有重要意義和潛力。本文于2023年8月發表在《Molecular Cancer》IF: 37.3期刊上
m6A修飾
m6A修飾是真核細胞mRNA和lncRNA中最常見的化學修飾。它幾乎參與了RNA的加工、核輸出、翻譯和降解等整個RNA代謝過程。RNA的m6A修飾是動態和可逆的。這種復雜的反應離不開“編寫者”、“擦除者”和“閱讀者”的互動。具體地說,m6A的“編寫者”和“擦除者”負責調節相關RNA的甲基化水平。這樣,各種m6A“閱讀者”就可以識別并結合m6A修飾的RNA來調節基因表達(圖1;表1)。
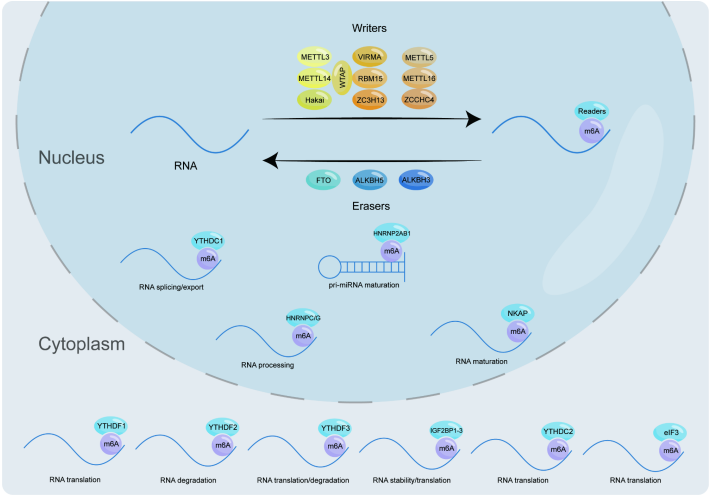
圖1 m6A修飾的調控
表1 m6A甲基化酶在RNA代謝中的功能
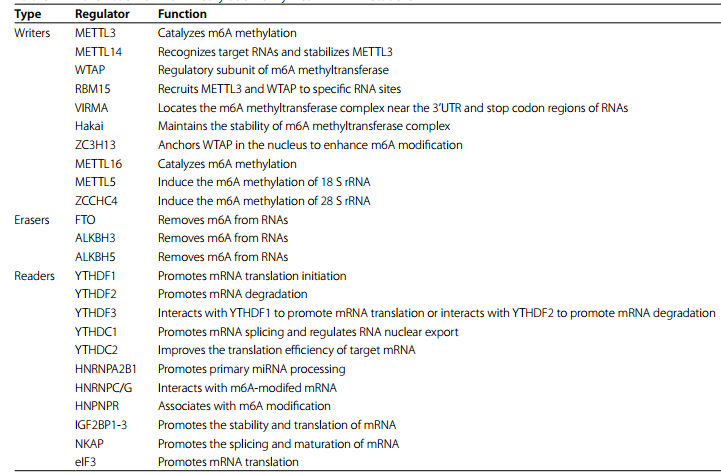
m6A“編寫者”- 甲基化轉移酶
作為m6A“編寫者”,甲基轉移酶復合物由多個成分組成,這些成分相互支持,共同調節核酸的甲基化。眾所周知,METTL3和METTL14是來自同一家族的S-腺苷甲硫氨酸(SAM)依賴性甲基轉移酶。它們形成的異二聚體在m6A的調節中起著至關重要的作用,同時也與體外DNA損傷的修復有關。這里,主要討論METTL3和METTL14作為m6A“編寫者”的作用。Wang等人證明,METTL3在分子水平上折疊形成帶正電溝槽,并含有大量的SAM,而在METTL14中沒有發現SAM。這說明METTL3是催化甲基化修飾的核心力量,但對METTL3的賴氨酸殘基進行類泛素修飾后,其催化能力受到抑制。此外, METTL3的功能也與Cys-Cys-Cys-His(CCCH)型鋅結合基序的結構基礎密不可分。作為METTL3-METTL14二聚體復合物的另一個組分,METTL14可以通過其末端RGG重復序列與RNA結合,也可以在分子水平上與METTL3形成廣泛的連接,從而提高METTL3的生物活性。METTL14還能在METTL3的保護下避免U-box-蛋白 1介導的泛素化降解。腎母細胞瘤1相關蛋白(WTAP)是催化m6A修飾的核心復合物的組分之一。它作為METTL3- METTL14復合體的調控亞基存在,其表達也受METTL3的調控。RNA結合基序蛋白15(RBM15)主要與METTL3和WTAP結合。它還通過促進RNA解旋酶DBP5與mRNA的結合來提高mRNA的成核效率。VIRMA在招募由METTL3、METTL14和WTAP組成的催化核心復合體方面發揮作用。它主要在RNA的3’非編碼區和終止密碼子附近發揮催化功能。Hakai是一種環指E3泛素連接酶,在維持m6A甲基轉移酶復合體的穩定性方面發揮著不可替代的作用。ZC3H13就像一顆鉚釘,連接著RBM15、WTAP和Virma,也與WTAP、Virma和Hakai的核定位有關。
除了上述,還有一些甲基轉移酶是不容忽視的。例如,METTL16是近年來逐漸被發現的一種甲基轉移酶,它通過其N-末端結構域和C-末端VCRs來調節MAT2A mRNA和U6 snRNA的表達,維持細胞內SAM的動態平衡,并與DNA損傷后的修復有關。此外,METTL5和ZCCHC4還負責細胞中18 S rRNA和28 S rRNA的甲基化。其作用的具體機制有待進一步研究。
M6A“擦除者”-去甲基化酶
脂肪量和肥胖相關蛋白(FTO)、ALKB同源物5(ALKBH5)和ALKB同源物3(ALKBH3)都是雙加氧酶ALKB蛋白家族的成員。它們依靠α-酮戊二酸和鐵(II)來降低RNA的m6A修飾水平。在微觀結構水平上,ALKBH3、ALKBH5和FTO都具有淺間隙,因此它們更喜歡與mRNA等單鏈核酸結合。此外,FTO的去甲基化活性受RNA序列和三級結構的影響,并且它還具有SFPQ(一種RNA結合蛋白)作為其伴侶蛋白。SFPQ可以與RNA上的CUGUG序列結合并募集FTO來促進近端去甲基化。
M6A“閱讀者”-甲基化閱讀蛋白
與“編寫者”和“擦除者”不同,m6A“閱讀者”不直接改變甲基化水平,而是識別并結合RNA的甲基化位點。在這個過程中,各種“閱讀者”與各種m6A位點結合,影響RNA的命運。
含有YT521-B同源(YTH)結構域的蛋白質YTHDF1-3是m6A修飾的常見“閱讀者”。一般認為YTHDF1可以誘導mRNA翻譯;YTHDF2加速mRNA的降解;YTHDF3具有以上兩個功能。但最近,一個新的YTHDF蛋白模型顯示,YTHDF1/2/3作用于相同的mRNAs子集,導致其降解,但不促進翻譯。該模型還表明,它們之間存在一定的功能補償。另一類具有YTH結構域的m6A“閱讀者”是YTH Domain Containing 1(YTHDC1)和YTHDC2,前者與RNA剪接和核輸出有關,后者與RNA翻譯有關。異質核核糖核蛋白(HNRNP)家族是另一個m6A“閱讀者”。其中,核內不均一核糖核蛋白A2/B1(HNRNPA2B1)可與含有RGm6AC序列的miRNA結合,募集microRNA微處理器復合物蛋白DGCR8剪接miRNA的前體,促進成熟miRNA的形成。HNRNPC和HNRNPG均與m6A開關的特殊機制有關。所謂m6A開關,是指m6A修飾的RNA結構發生變化,影響RNA結合蛋白與相應位點的結合,從而調控細胞內相關生命活動的過程。最近研究表明,HNPNPR也是HNRNPs蛋白家族的一員,與m6A修飾有很強的相關性,參與腫瘤糖酵解。此外,胰島素樣生長因子2 mRNA結合蛋白(igf2bp)主要參與調控mRNA的穩定性和翻譯。NF-κB激活蛋白(NKAP)促進mRNA的剪接和成熟。真核生物翻譯起始因子3(Eukaryotic translation initiation factor 3, eIF3)也被認為是m6A的“讀者”,在真核生物翻譯起始過程中起核心作用。由此可見,m6A“讀者”所起的作用是非常復雜多樣的,包括但不限于RNA剪接、成熟、穩定、翻譯、定位等調控。在這方面,還有許多未知的“讀者”等待被發現。
糖酵解過程
糖酵解是將一分子葡萄糖分解成兩分子丙酮酸,然后產生兩分子ATP的過程。在這個過程中,胞外葡萄糖在葡萄糖轉運蛋白(GLUT)的幫助下轉移到細胞液中。葡萄糖在己糖激酶(HK)、葡萄糖-6-磷酸異構酶(GPI)、磷酸果糖激酶-1(PFK)和醛縮酶(ALD)的作用下分解為相互轉化的甘油醛- 3-磷酸和二羥丙酮磷酸。到目前為止,含有六個碳原子的葡萄糖已經分解成兩個三碳單元。然后,在甘油醛-3-磷酸脫氫酶(GAPDH)、磷酸甘油酸激酶(PGK)、磷酸甘油酸變化酶(PGAM)、烯醇化酶(ENO)和丙酮酸激酶(PK)的催化下,最終轉化為兩分子丙酮酸。
當細胞為需氧且有線粒體時,丙酮酸通常進入線粒體進行三羧酸循環,產生大量ATP,為細胞提供能量;然而,如果細胞缺氧或缺乏線粒體,丙酮酸就會被還原成乳酸。在這些過程中,代謝中間體也可以進入其他生化反應過程,使細胞內的各種反應得以協調有序。然而,生物化學家Otto Warburg發現了一個特殊的現象:在癌細胞中,即使它們有正常的線粒體和充足的氧氣,它們也很少發生氧化磷酸化(OXPHOS)。相反,癌細胞主要依靠糖酵解提供能量。這一特性表現為癌細胞能夠耐受低氧環境,同時也增加了腫瘤細胞微環境中的乳酸水平,刺激腫瘤生長發育。
糖酵解與腫瘤
對于大多數正常細胞來說,它們更喜歡氧化磷酸化為自己提供能量。然而,在腫瘤細胞中,糖酵解具有獨特的地位。這是因為與OXPHOS相比,雖然糖酵解產生的ATP少,但速度更快。因此,糖酵解同時可以產生更多的ATP,為腫瘤細胞的生命活動提供能量。此外,糖酵解過程中產生的各種中間產物對于細胞內脂質、核酸等生物大分子的合成至關重要,有助于腫瘤細胞的增殖。乳酸是糖酵解的產物之一,能有效抑制多種免疫細胞的功能,有助于腫瘤細胞的免疫逃逸。近年來,越來越多的研究證實,腫瘤糖酵解在腫瘤增殖、血管生成和淋巴管生成、轉移、免疫逃逸等腫瘤進展過程中起著不可或缺的作用,并與腫瘤預后評價和治療有一定的相關性(圖2;表2)。

圖2 糖酵解與腫瘤的關系
表2 糖酵解在腫瘤發生發展中的作用
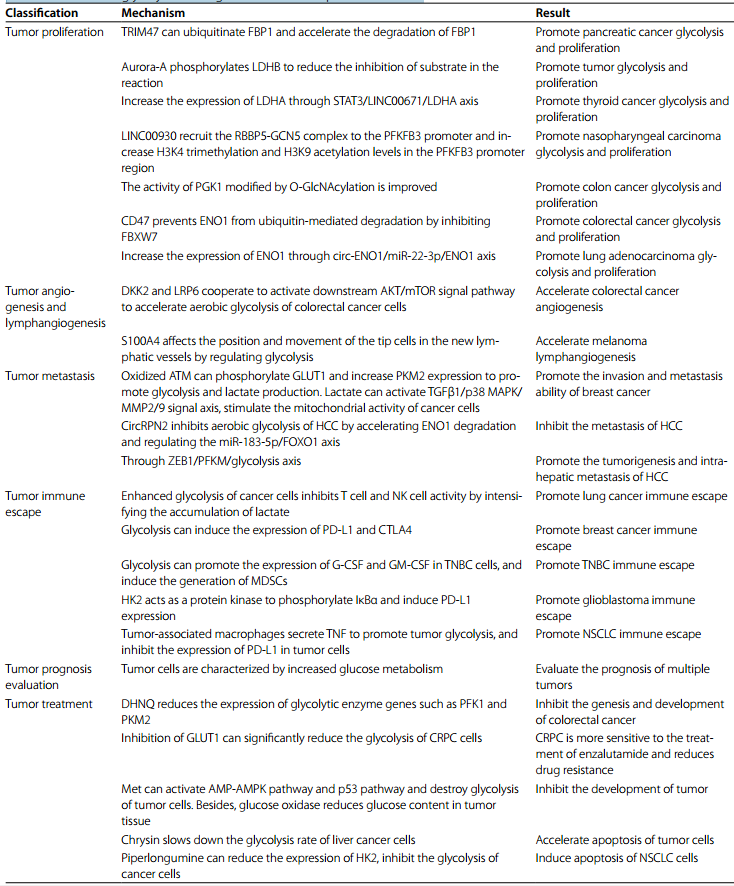
糖酵解與腫瘤增殖
細胞的生命活動離不開能量。如果沒有糖酵解提供能量,即使是頑強的腫瘤細胞也無法存活。在胰腺癌中,TRIM47可以泛素化果糖-1,6-二磷酸酶(FBP1),并加速FBP1的降解,從而促進胰腺癌細胞的糖酵解和增殖。
LDH是催化丙酮酸還原為乳酸正向反應的關鍵酶,LDHB是LDH的一個亞基。絲氨酸/蘇氨酸激酶Aurora-A可以直接結合乳酸脫氫酶B(LDHB)并使其絲氨酸162位點磷酸化。磷酸化的LDHB降低了底物在還原反應中的抑制作用,顯著加速丙酮酸向乳酸的轉化,促進糖酵解和腫瘤細胞的增殖。除LDHB外,LDHA作為LDH的組成部分,在腫瘤增殖中也起著重要作用。在甲狀腺癌中,lncRNA LINC00671與LDHA水平呈負相關。在缺氧條件下,轉錄因子STAT3被激活,抑制LINC00671的表達。這增加了腫瘤細胞中的LDHA含量,促進糖酵解和甲狀腺癌的增殖。
果糖-2,6-二磷酸酶3(PFKFB3)是糖酵解的調節酶。在LINC00930的作用下,更多的RBBP5-GCN5復合物與PFKFB3啟動子結合。因此,PFKFB3啟動子區域的H3K4三甲基化和H3K9乙酰化增加,導致PFKFB3的表達顯著增加。過表達的PFKFB3可以催化乳酸的產生,調節細胞周期相關蛋白的表達,促進鼻咽癌細胞糖酵解和增殖。
作為糖酵解過程中的關鍵酶,PGK1催化1,3-二磷酸甘油酸轉化為3-磷酸甘油酸,并產生一個ATP分子。在結直腸癌中,PGK1中的O-連接N-乙酰氨基葡萄糖(O-GlcNAc)修飾蘇氨酸255位的糖基化位點。經O-GlcN酰化修飾的PGK1活性大大提高,并能在線粒體外膜轉位酶TOM20的幫助下進入線粒體。進入線粒體的PKG1可以抑制癌細胞中的OXPHOS,但可以提高糖酵解活性和乳酸的產生。因此,結腸癌細胞通過提高PGK1 O-GlcN酰化水平促進腫瘤增殖酰化水平促進腫瘤增殖。
ENO1能催化2-磷酸甘油酸生成磷酸烯醇式丙酮酸。研究表明,CD47的過度表達與結直腸癌的預后不良有關。從機制上講,CD47通過抑制FBXW7來阻止ENO1泛素介導的降解。這可以增強腫瘤細胞的糖酵解,激活ERK信號通路,最終加速腫瘤細胞的增殖。在肺腺癌中,CIRC-ENO1通過與miR-22-3p相互作用上調ENO1基因的表達,導致腫瘤細胞的ATP水平、葡萄糖攝取和乳酸生成增加,從而促進腫瘤的增殖。
糖酵解與腫瘤血管生成和淋巴管生成
腫瘤血管生成和淋巴管生成是一個復雜的過程。這一過程的異常是腫瘤轉移的重要特征之一。Dickkopf相關蛋白2(DKK2)可與脂蛋白受體相關蛋白6(LRP6)協同作用,激活下游的AKT/mTOR信號通路,促進結直腸癌細胞的有氧糖酵解。此時,糖酵解產生的大量乳酸在腫瘤微環境中積聚,進一步刺激內皮細胞生長,加速腫瘤血管生成和結直腸癌的轉移。
腫瘤淋巴管生成和前哨淋巴結轉移是腫瘤進展的關鍵。Li等通過對黑色素瘤淋巴結轉移小鼠模型的研究發現,S100A4在小鼠體內的表達可以增強淋巴管內皮細胞(LECs)的運動能力,促進黑色素瘤淋巴管生成和淋巴結轉移。在低氧條件下,LECs中S100A4的表達上調,激活AMPK依賴的糖酵解,并通過影響腫瘤淋巴管尖端細胞在新淋巴管中的位置和運動來調節腫瘤淋巴管的出芽。
糖酵解與腫瘤轉移
在腫瘤微環境中,癌相關成纖維細胞(cancer-associated fibroblasts, CAF)是大量能夠分泌多種癌細胞調節因子的基質細胞。CAF分泌的調節因子能促進腫瘤的發生發展。CAF還具有很強的糖酵解能力,在細胞外釋放大量乳酸。之后,這部分乳酸會被癌細胞吸收,在癌細胞漿中轉化為丙酮酸,然后通過線粒體的OXPHOS為癌細胞的增殖提供持續的能量。這個過程被稱為“reverse Warburg effect”。腫瘤相關成纖維細胞中的共濟失調-毛細血管擴張突變蛋白激酶(ATM)在低氧條件下可被誘導氧化。氧化ATM可使GLUT1磷酸化,增加PKM2的表達,促進糖酵解。在這個過程中,大量積累的乳酸可以激活轉化生長因子β1/p38MAPK/MMP2/9信號軸,刺激癌細胞的線粒體活性,提高乳腺癌細胞的侵襲和轉移能力。
Li等人的動物模型顯示,CircRPN2抑制肝細胞癌的糖酵解和轉移。具體地說,CircRPN2通過加速ENO1的降解和調節miR-183-5p/FOXO1軸來抑制肝癌的有氧糖酵解,最終抑制肝癌的轉移。此外,周和他的同事分析了來自TCGA數據庫的數據,發現ZEB1-PFKM-糖酵解軸與肝癌的腫瘤發生和肝內轉移顯著相關。此外,在原位肝細胞癌移植瘤的隨訪實驗中也得到了相同的結果。
糖酵解與腫瘤免疫逃逸
腫瘤的免疫逃逸是指癌細胞逃避免疫系統的監視和攻擊,從而在體內存活和增殖的現象。大量證據表明,腫瘤糖酵解與多種癌細胞的免疫逃逸密切相關。研究發現,Notch1信號與TAZ形成的正反饋環具有促進糖酵解基因表達,加速癌細胞乳酸合成的作用。胞外液中積累的乳酸可抑制T、NK細胞活性,誘導肺癌免疫逃逸。在乳腺癌中,糖酵解活性升高可上調IL-17信號通路,募集巨噬細胞等免疫細胞,但降低了腫瘤殺傷細胞的聚集。高糖酵解還可誘導PD-L1、CTLA-4等免疫檢查點的表達,使乳腺癌逃脫免疫系統的監視。此外,腫瘤糖酵解還可促進三陰性乳腺癌(TNBC)細胞中粒細胞集落刺激因子(G-CSF)和粒細胞巨噬細胞集落刺激因子(GM-CSF)的表達,誘導腫瘤髓系抑制細胞(MDSCs)的產生,影響腫瘤免疫逃逸和腫瘤生長。
在膠質母細胞瘤中,高水平的有氧糖酵解將HK2從線粒體轉移到細胞質。然后,HK2作為一種蛋白激酶,磷酸化IκBα,誘導PD-L1表達,導致腫瘤免疫逃逸。此外,研究發現,腫瘤相關巨噬細胞(TAM)通過減少腫瘤免疫微環境中的氧含量,分泌腫瘤壞死因子促進腫瘤糖酵解,抑制腫瘤細胞中PD-L1的表達,參與非小細胞肺癌的糖酵解和免疫逃逸。
糖酵解與腫瘤預后評估
18F-FDG PET/CT顯像是一種無創性評估各種惡性腫瘤的分期、復發或治療反應的工具。這項檢查最常用的顯像劑是18F-FDG,它是一種葡萄糖類似物。被注射到人體后,它可以像葡萄糖一樣進入細胞,被磷酸化形成FDG-6-磷酸。然而,它不能進一步代謝,只能在細胞內積累。腫瘤細胞的特征是糖代謝增加,表現為葡萄糖攝取和代謝率增加。因此,PET/CT的成像原理是利用18F-FDG在各種惡性腫瘤中的高代謝、高攝取和蓄積來發現原發灶和轉移灶。
目前,結合腫瘤體積和腫瘤代謝活性的18F-FDG PET/CT參數,如代謝腫瘤體積和總病變糖酵解,是預測多種癌癥的預后成像生物標志物,如口咽鱗狀細胞癌、上皮性卵巢癌、腎細胞癌和結直腸癌。
糖酵解與腫瘤治療
腫瘤細胞為了適應惡劣的腫瘤微環境,通過調節有氧糖酵解為其各種生命活動提供能量支持。因此,破壞腫瘤細胞糖酵解被認為是一種潛在的腫瘤治療策略。目前,已開發出幾種靶向糖酵解的抑制劑,但仍需進一步研究,使其發揮更全面的腫瘤抑制作用。
DHNQ是一種新的磷脂酰肌醇3-激酶(PI3K)信號通路抑制劑。DHNQ通過減少PFK1、PKM2等糖酵解酶基因的表達,阻斷癌細胞的糖酵解,從而抑制結直腸癌的增殖、遷移和血管生成。在去勢抵抗前列腺癌(CRPC)細胞中,雄激素受體直接與GLUT1基因啟動子結合,促進GLUT1轉錄,以滿足CRPC對葡萄糖的大量需求。抑制GLUT1可以顯著降低CRPC細胞的糖酵解和增殖率,使CRPC對苯扎魯胺(藥名)的治療更加敏感,并降低耐藥性。
二甲雙胍是一種選擇性抑制HK2活性的藥物。它通過激活AMP-AMPK通路和P53通路,破壞腫瘤細胞的糖酵解。葡萄糖氧化酶通過消耗積聚的葡萄糖來降低腫瘤組織中葡萄糖的含量,從而提高饑餓治療的效果。兩種藥物聯合用藥的抑瘤效果優于單獨用藥。
除了上述抑制劑外,還有一些天然化合物可以調節腫瘤的糖酵解和增殖。徐等人發現,生物活性黃酮白楊素通過減少HK2的表達,減緩肝癌細胞的糖酵解速度,加速肝癌細胞的凋亡。胡椒堿是另一種具有抗腫瘤作用的天然化合物。在非小細胞肺癌中,胡椒堿可以降低HK2的表達,抑制癌細胞的糖酵解,促進癌細胞的凋亡,達到抗腫瘤的作用。
m6A調節腫瘤糖酵解
越來越多的證據表明,m6A修飾通過多種途徑調節腫瘤糖酵解,對腫瘤的增殖、轉移和治療具有重要意義(圖3;表3)。
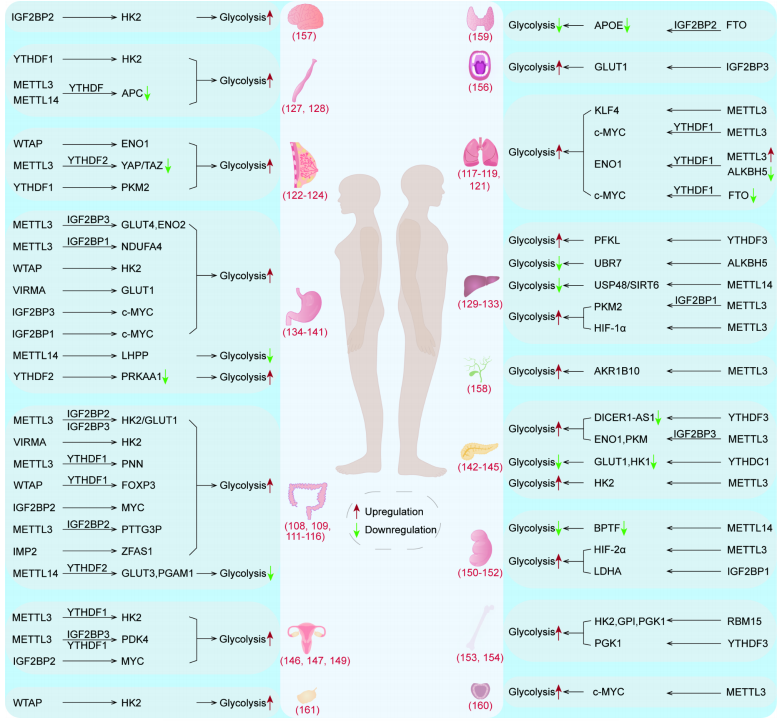
圖3 m6A調節腫瘤糖酵解
表3 腫瘤糖酵解中的m6A甲基化

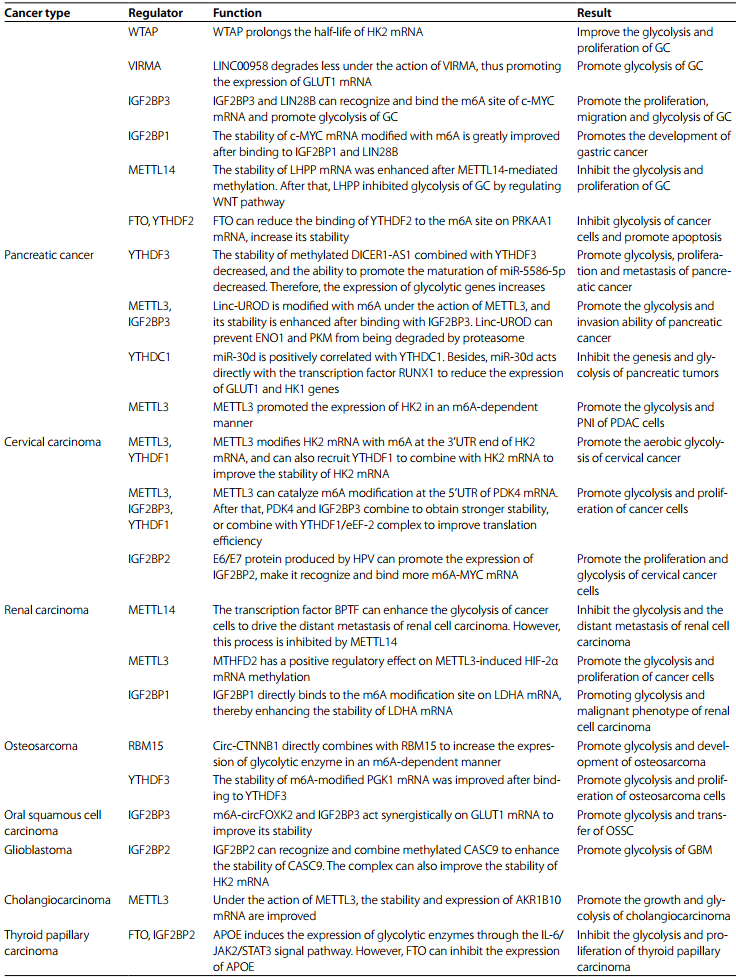

結直腸癌
結直腸癌(CRC)是全球第二大癌癥死亡原因。在結直腸癌細胞中,METTL3直接作用于HK2和GLUT1基因,M6A修飾的HK2和GLUT1基因的穩定性增強。在隨后的過程中,IGF2BP2與HK2的5’/3’非編碼區結合。IGF2BP2/3與GLUT1的3’非編碼區結合。這促進了HK2和GLUT1的表達,促進結直腸癌細胞的糖酵解和增殖。另一種甲基轉移酶VIRMA以m6A依賴的方式增加HK2 mRNA的甲基化水平,上調HK2 mRNA的水平并提高其mRNA穩定性,最終可加速癌細胞的有氧糖酵解,提高腫瘤的惡性程度。Liu等人的分析表明,m6A可以修飾GLUT1基因,增強GLUT1基因的穩定性,從而促進結直腸癌的糖酵解和細胞增殖。Pinin(PNN)是一種橋粒相關蛋白,參與了一些腫瘤的發生發展。He等人發現PNN mRNA在METTL3的作用下經歷了m6A修飾,并通過與YTHDF1結合來提高穩定性,最終促進了結腸癌的糖酵解和增殖。Zhang等人發現WTAP和YTHDF1以m6A依賴的方式增強FOXP3mRNA的穩定性。FOXP3的高表達可直接促進下游糖酵解過程,也可通過上調SMARCE1的表達促進結腸腺癌的糖酵解和增殖。除了直接調節糖酵解相關酶的mRNA外,M6A修飾還可以作用于一些lncRNAs和miRNAs來調節腫瘤的糖酵解。Wang等人發現IGF2BP2穩定性的長基因間非編碼RNA(LINRIS)在結直腸癌細胞中處于高水平,并與患者的預后不良有關。LINRIS可以誘導IGF2BP2的K139泛素化,從而阻止溶酶體對IGF2BP2的降解,使IGF2BP2蛋白的含量保持在一定水平,并通過IGF2BP2/MYC/糖酵解軸促進癌細胞的糖酵解和增殖。鄭等人的研究表明,lncRNA PTTG3P的表達與結直腸癌患者的預后密切相關。PTTG3P在METTL3的作用下甲基化,并與IGF2BP2結合以獲得更高的穩定性。之后,在轉錄調控因子YAP1的協同作用下,促進了結直腸癌的糖酵解和增殖。作為m6A的“閱讀者”,IMP2通過HH3-4結構域與m6A修飾的LncRNA ZFAS1相互作用,增加了ZFAS1的穩定性。高表達的ZFAS1可以與Obg-like ATPase 1(OLA1)結合,提高其水解三磷酸腺苷和激活糖酵解的能力。通過對表達野生型p53的CRC細胞的研究,侯和他的同事發現,野生型p53作用于METTL14的啟動子區域,并誘導METTL14的表達。其次,METTL14在YTHDF2的輔助下,促進了pri-miR-6769b和pri-miR-499a的生物成熟。最終,MiR-6769b-3p/GLUT3軸和miR-499a-3p/PGAM1軸通過降低GLUT3和PGAM1的表達,抑制p53 WT結直腸癌細胞的有氧糖酵解和惡性表型。
肺癌
肺癌是世界上第二常見的癌癥。在非小細胞肺癌中,經METTL3介導的m6A修飾后,lncRNA ABHD11-AS1更加穩定,并通過ABHD11-AS1/EZH2/KLF4軸促進非小細胞肺癌的有氧糖酵解。張和他的同事們發現,METTL3還可以增加lncRNA DLGAP1-AS2的甲基化水平,并提高其穩定性。接下來,DLGAP1AS2和YTHDF1共同促進c-MYC mRNA的表達,加速癌細胞的生長,促進糖酵解。
在肺腺癌(LUAD)中,ENO1以M6A依賴的方式促進腫瘤的發展。在肺腺癌(LUAD)中,ENO1以M6A依賴的方式促進腫瘤的發展。肺腺癌組織中METTL3基因表達上調,ALKBH5基因表達下調,M6A水平和ENO1基因甲基化水平升高。YTHDF1,一個m6A“閱讀者”,與甲基化的ENO1 mRNA相結合,導致ENO1表達增加,并促進肺腺癌的糖酵解。劉和他的同事通過分析TCGA和GEO數據集發現,NPM1與m6A修飾和糖酵解有關。他們還認為,m6A修飾可能通過增強NPM1的穩定性來促進LUAD中ENO1、HK2、LDHA、LDHB和GLUT1等糖酵解酶的表達,從而增強LUAD的糖酵解能力和發。Yang等發現WnT/β-連環素軸可以作用于FTO的啟動子區域,從而抑制FTO的表達。增加c-MYC基因m6A的修飾,募集YTHDF1促進c-MYC基因的翻譯,最終加速肺腺癌細胞的糖酵解和增殖。
乳腺癌
到目前為止,乳腺癌已經超過肺癌,成為世界上最常見的癌癥。在乳腺癌中,C5aR1陽性的中性粒細胞可以促進乳腺癌細胞的糖酵解。從機制上講,C5aR1陽性中性粒細胞分泌的白介素1β和腫瘤壞死因子α可作用于下游的ERK1/2-WTAP-ENO1信號軸,增加eNO1mRNA的m6A修飾水平,促進乳腺癌細胞中eNO1的表達和糖酵解。徐和他的同事們發現,m6A還可以通過Hippo途徑調節乳腺癌的糖酵解。具體地說,METTL3/LATS1/YTHDF2軸通過抑制河馬途徑中的YAP/TAZ促進乳腺癌的糖酵解和腫瘤發生。姚等人發現,在低氧條件下,腫瘤細胞中HIF-1α轉錄增加,miR-16-5p的表達受到抑制,導致miR-16-5p與YTHDF1的結合減少,促進YTHDF1的表達。YTHDF1隨后與PKM2 mRNA結合,增加癌細胞中PKM2的表達和糖酵解,加速乳腺癌的發生和轉移。此外,生物信息學分析表明,GPI與乳腺癌的m6A修飾密切相關,并可能成為乳腺癌的一個新的預后標志物。
食道癌
目前,食道癌的總死亡率和發病率分別排在第六位和第七位。TCGA ESCA隊列分析表明,關鍵糖酵解酶HK2與食道癌M6A修飾密切相關。功能研究表明,人類白細胞抗原復合體P5(HCP5)可增強YTHDF1與m6A修飾的HK2 mRNA的結合,從而提高HK2mRNA的穩定性,促進ESCC細胞的有氧糖酵解,增加ESCC的惡性表型。
APC是一種常見的抑癌基因。在食道癌中,METTL3和METTL14協同作用于APC mRNA,M6A修飾的APC mRNA與YTHDF結合后降解。因此,APC對Wnt/β-catenin通路的調節作用減弱,細胞周期蛋白D1、c-myc和PKM2的表達增加,從而促進腫瘤的有氧糖酵解和腫瘤的發展。
肝癌
肝癌是世界上第六大常見癌癥,但它是導致癌癥死亡的第三大原因。在肝細胞癌中,作為m6A“閱讀者”的YTHDF3抑制了m6A修飾的PFKL mRNA的降解,促進了PFKL的表達和有氧糖酵解。令人驚訝的是,PFKL正向調節YTHDF3蛋白的表達,形成YTHDF3-PFKL-YTHDF3的正反饋循環,促進肝癌的生長和肺轉移。趙等人的研究表明,泛素蛋白連接酶E3組分N-識別素7(UBR7)激活Keap1/Nrf2/Bach1/HK2軸,減少肝癌細胞中HK2的含量,抑制肝癌細胞的糖酵解和增殖。然而,m6A“擦除器”ALKBH5以m6A依賴的方式促進UBR7的表達。Du等發現METTL14/USP48/SIRT6軸也具有抑制肝癌的作用。這是因為肝癌細胞中的泛素特異肽酶48(USP48)可以與組蛋白脫乙酰基酶SIRT6結合,提高SIRT6的穩定性,從而起到削弱肝癌細胞糖酵解的作用。USP48也受METTL14監管。
LNCAROD是一種與肝細胞癌糖酵解和預后不良相關的lncRNA。在METTL3和IGF2BP1的作用下,LNCAROD的表達和穩定性都有所提高。LNCAROD的高表達可促進SRSF3介導的PKM向PKM2的轉化,并可作為miR-145-5p的ceRNA,維持胞漿內PKM2的水平。這兩條途徑協同增加PKM2的水平,誘導肝癌細胞的有氧糖酵解、增殖和侵襲。楊和他的同事在他們對乙肝病毒X相互作用蛋白(HBXIP)的研究中發現,HBXIP的表達可以促進肝癌細胞的糖酵解,提高腫瘤的惡性程度。這是因為HBXIP可以誘導甲基轉移酶METTL3的表達。在METTL3過表達的肝癌細胞中,HIF-1α的m6A修飾水平增加,激活下游糖酵解酶,增加肝癌的侵襲能力。
胃癌
胃癌的死亡率和發病率分別位居第四和第五位。在胃癌中,m6A在METTL3的作用下修飾HDGF mRNA,與IGF2BP3結合后,其穩定性和表達增強。細胞核內的HDGF可與GLUT4和ENO2啟動子結合,增加細胞內糖酵解酶的含量,促進胃癌的糖酵解、增殖和肝轉移METTL3還可以催化NADH脫氫酶-1α亞復合物4(NDUFA4)mRNA 3’UTR的m6A修飾,募集IGF2BP1增加NDUFA4 mRNA的穩定性,促進NDUFA 4表達。NDUFA4作為線粒體電子傳遞鏈復合體的組成成分,通過促進GC細胞糖酵解而促進胃癌細胞增殖和腫瘤生長。另一種甲基轉移酶WTAP催化HK2 mRNA 3’UTR的m6A修飾,延長HK2 mRNA的半衰期,并促進GC細胞的糖酵解和增殖。
LncRNA和WNT通路在胃癌糖酵解中也起重要作用。比如LINC00958可以促進GC細胞的有氧糖酵解。在機制上,m6A甲基轉移酶VIRMA誘導LINC00958甲基化位點的m6A修飾。m6A修飾后的LINC00958降解減少,并與GLUT1 mRNA結合,提高GLUT1 mRNA的穩定性,從而加速GC的有氧糖酵解。徐等發現RNA結合蛋白IGF2BP3和LIN28B可以識別并結合c-MYC mRNA的m6A位點。LncRNA LOC101929709作為一個支架來支撐以上三者的結合。c-MYC的過表達反過來促進LOC101929709和LIN28B的表達,形成一個正反饋環,促進胃癌的增殖、遷移和糖酵解。此外,羅等發現IGF2BP1的過表達與胃癌的不良預后有關。機制上,m6A修飾的c-MYC mRNA與IGF2BP1結合后穩定性大大提高。c-MYC的過度表達通過c-MYC/糖酵解軸促進胃癌的發展。
與上述方式不同,METTL14介導的甲基化后,磷酸賴氨酸磷酸組氨酸無機焦磷酸磷酸酶(LHPP)mRNA的穩定性增強,起到抑制胃癌糖酵解的作用。之后LHPP的乙酰化可以抑制GSK3b的磷酸化進而抑制WNT途徑,從而抑制胃癌細胞的糖酵解和增殖。在Zhang等人的另一項研究中,發現FTO去除了催化亞基α1(prkaa 1)mRNA 3’UTR區的m6A修飾,阻止了YTHDF2識別和結合PRKAA1 mRNA。結果,PRKAA1 mRNA的穩定性增加,最終促進胃癌細胞中的糖酵解并抑制凋亡。
m6A調節腫瘤糖酵解的臨床意義
上述信息表明,m6A調節的腫瘤糖酵解在腫瘤發生和發展中起重要作用。這也引起了研究者對靶向m6A調控的腫瘤糖酵解是否有利于腫瘤治療的好奇。因此,在這里,進一步討論m6A調控的腫瘤糖酵解在癌癥發病機制、藥物治療反應和耐藥性中的作用機制,以及靶向m6A調控的腫瘤糖酵解的治療潛力(圖4;表4)。
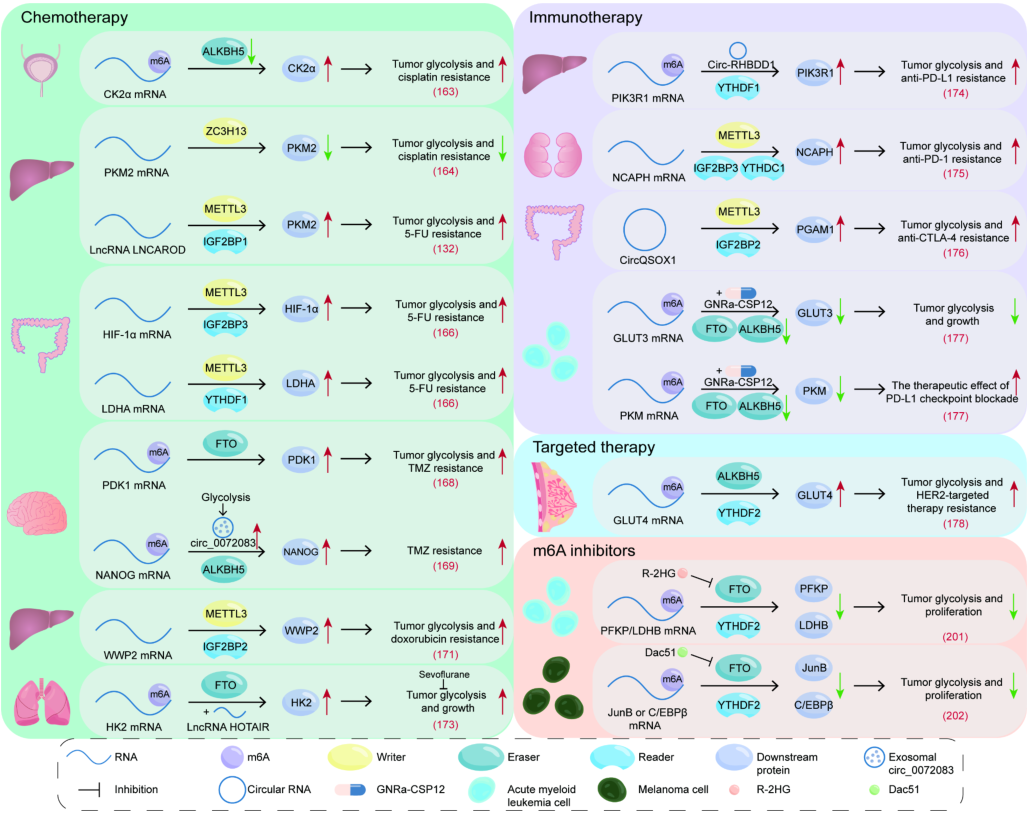
圖4 m6a調節腫瘤糖酵解的臨床意義。m6A調節通過影響糖酵解影響腫瘤化療、免疫治療和靶向治療的療效。此外,m6A抑制劑通過抑制糖酵解發揮抗腫瘤作用
表4 m6A通過調節腫瘤糖酵解影響腫瘤治療
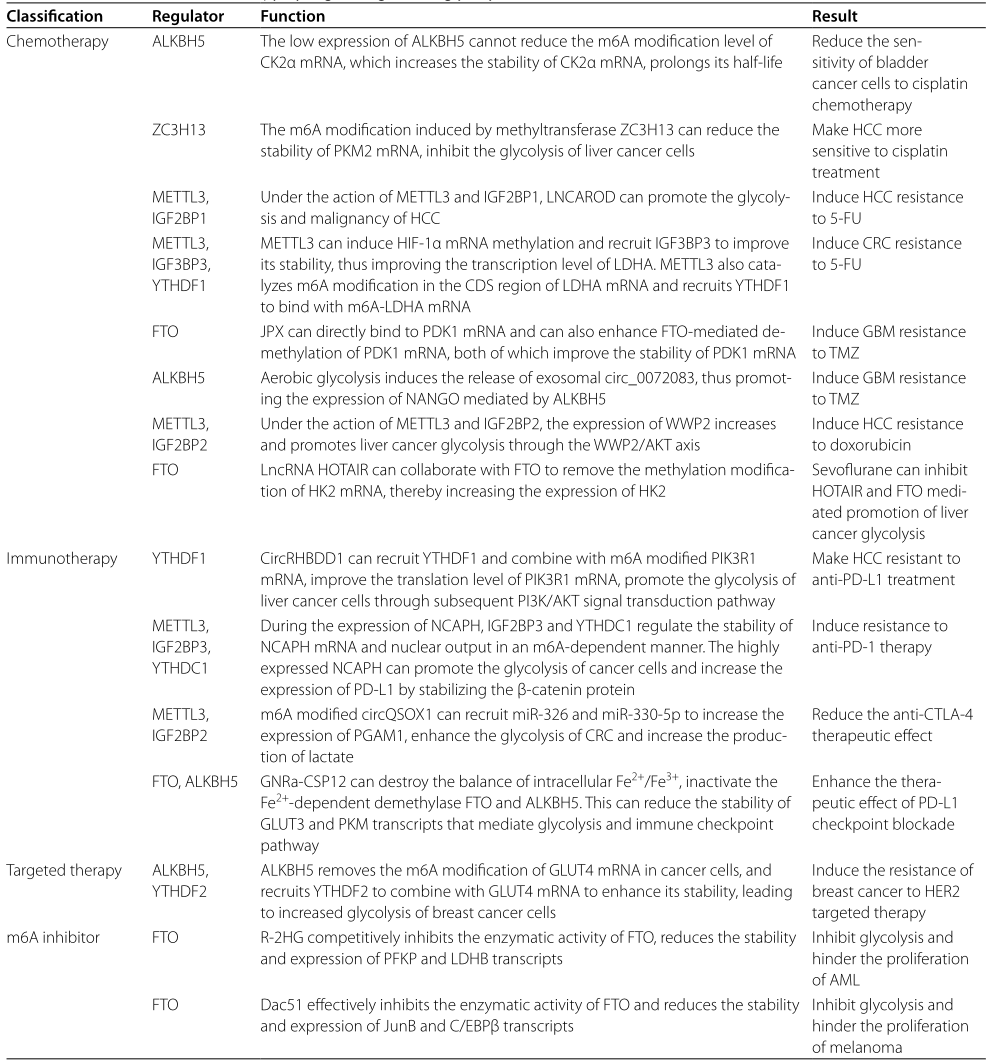
在腫瘤化療中
癌癥的化療是通過使用某些化學藥物破壞或抑制癌細胞的增殖來治療癌癥的方法。一些研究表明,腫瘤糖酵解中的m6A修飾與對化療的反應有關。順鉑可以抑制癌細胞分裂,誘導癌細胞死亡。因此,它已成為治療多種實體腫瘤的有效化療藥物。在膀胱癌組織中,ALKBH5的低表達不能降低CK2α mRNA的m6A修飾水平,這增加了CK2α mRNA的穩定性,延長了其半衰期,最終加速了膀胱癌細胞的糖酵解、增殖、遷移和侵襲,降低了膀胱癌細胞對順鉑化療的敏感性。Wang等在對肝癌的研究中發現,甲基轉移酶ZC3H13誘導的m6A修飾可以降低PKM2 mRNA的穩定性,抑制肝癌細胞的糖酵解,降低惡性程度,并使肝癌對順鉑治療更加敏感。
在腫瘤免疫治療中
腫瘤免疫治療是通過重新啟動和維持腫瘤免疫循環,恢復機體正常的抗腫瘤免疫反應。目前,m6A修飾的腫瘤糖酵解在腫瘤免疫檢查點抑制劑(抗PD-L1/抗PD-1和抗CTLA-4)治療中的作用已引起人們的關注。
在肝癌中,CircRHBDD1可以募集YTHDF1并與m6A修飾的PIK3R1mRNA結合,提高PIK3R1mRNA的翻譯水平,通過隨后的PI3K/AKT信號轉導途徑促進肝癌細胞的糖酵解,使肝癌對抗PD-1治療產生耐藥。因此,糖酵解抑制劑和免疫抑制劑聯合治療肝癌值得關注。Chen等人發現非SMC凝集素I復合體H(NCAPH)與腎透明細胞癌的糖酵解和免疫耐受密切相關。在NCAPH表達過程中,IGF2BP3和YTHDC1以m6A依賴的方式調節NCAPH mRNA的穩定性和核輸出。高表達的NCAPH通過穩定β-連環蛋白,促進腫瘤細胞糖酵解,增加PD-L1的表達,誘導抗PD-1治療耐藥。在結直腸癌中,CircQSOX1在METTL3的作用下發生甲基化,與IGF2BP2結合后其穩定性提高。m6A修飾的CircQSOX1可以募集miR-326和miR-330-5p增加PGAM1的表達,增強結直腸癌的糖酵解,增加乳酸的產生,促進結直腸癌的免疫逃逸,降低抗CTLA-4的治療效果。由此可見,sh-CircQSOX1與抗CTLA-4聯合治療對于避免結直腸癌免疫治療的耐藥性具有重要價值。此外,GNRa-CSP12是一種潛在的治療白血病的免疫抑制劑。它能破壞細胞內Fe2+/Fe3+的平衡,使依賴Fe2+的脫甲基酶FTO和ALKBH5失活,并增加細胞內m6A的水平。這可以降低分別介導糖酵解和免疫檢查點途徑的GLUT3和PKM轉錄物的穩定性,從而抑制AML細胞的增殖并增強PD-L1檢查點阻斷的治療效果。
在腫瘤靶向治療中
除了化療和免疫治療外,m6A的修飾還可能通過調節糖酵解來降低HER2靶向治療乳腺癌的療效。具體來說,ALKBH5去除癌細胞中GLUT4 mRNA的m6A修飾,并招募YTHDF2與GLUT4 mRNA結合以增強其穩定性,導致乳腺癌細胞糖酵解增加,并對HER2靶向治療產生耐藥性。因此,抑制ALKBH5/GLUT4軸對提高包括曲妥珠單抗和拉帕替尼在內的HER2靶向治療乳腺癌的療效具有重要意義。
聯合m6A和糖酵解抑制劑治療腫瘤
如前所述,m6A調控的腫瘤糖酵解在腫瘤的化療、免疫治療和靶向治療中具有不可估量的作用。這也提出了m6A和糖酵解抑制劑聯合使用是否有助于更好的抗腫瘤治療的問題。
為了滿足對葡萄糖的需求,絕大多數癌細胞的細胞膜上都有GLUT1的高表達。GLUT1特異性抑制劑,如STF31和WZB117,已被開發用于阻斷癌細胞對葡萄糖的攝取,從而抑制腫瘤生長。此外,針對糖酵解過程中關鍵酶的幾種抑制劑已被研究,包括2-脫氧-d -葡萄糖(2-DG)、PFK158、3-BrPA、紫草素和核黃素。這些抑制劑分別作用于HK2、PFKFB3、GAPDH、PKM2和LDH,有效抑制糖酵解和腫瘤增殖。其中,靶向HK2的糖酵解抑制劑2-DG已進入臨床試驗階段。
隨著高通量篩選技術的進步和對m6A修飾分子機制的不斷探索,出現了幾種具有抗癌潛力的m6A調節抑制劑。例如,STM2457和Elvitegravir是METTL3的特異性抑制劑,而CWI1-2和JX5選擇性抑制IGF2BP2。然而,FTO抑制劑的效果最為顯著。Rhein、FB23、FB23-2、MO-I-500、MA2、CS1和CS2均通過抑制FTO的m6A去甲基化酶活性顯示出良好的抗腫瘤作用。值得注意的是,這里重點介紹了兩種FTO抑制劑,R-2-羥基戊二酸(R-2HG)和Dac51。R-2HG競爭性地抑制FTO的酶活性,導致AML細胞中pFKP和LDHB的m6A修飾增加。隨后,YTHDF2識別并結合這些轉錄本,降低它們的穩定性和表達,最終通過抑制糖酵解途徑來阻礙AML的增殖。在黑色素瘤中,Dac51通過抑制FTO增強JunB和C/EBPβ mRNA的m6A修飾水平,這促進了YTHDF2與這兩種mRNA的結合,導致JunB和C/EBPβ表達顯著降低。JunB和C/EBPβ都是轉錄因子,能激活參與糖酵解的基因,如pFKP、PGAM1和HK1,從而促進腫瘤細胞的糖酵解,限制CD8+T細胞的激活和效應狀態,促進腫瘤的發生和發展。通過靶向這些過程,FTO抑制劑Dac51有效地抑制了黑色素瘤。有趣的是,在FTO-KD細胞中過表達JunB足以抑制CD8+T細胞的激活。用HK2抑制劑2-DG阻斷腫瘤糖酵解可顯著恢復受損的CD8+T細胞活化。
現有證據有力地表明,m6A可以通過調節腫瘤糖酵解調節各種癌細胞類型對抗癌治療的敏感性。因此,m6A抑制劑與腫瘤糖酵解抑制劑聯合使用有望提高抗腫瘤療效。人們熱切期待這些抑制劑在臨床應用上的早期突破,因為它們為未來的癌癥治療帶來了希望。
結論與未來展望
腫瘤和糖酵解之間的故事已經被探索了幾十年。最典型的例子是Warburg效應的發現,它從葡萄糖代謝的角度揭示了腫瘤細胞與正常細胞之間的差異。隨著MeRIP-seq、miCLIP、SELECT-m6A等m6A檢測技術的快速發展,對m6A與腫瘤糖酵解關系的研究越來越多。研究者發現m6A修飾通過一系列直接或間接的方式調節腫瘤細胞的糖酵解和增殖。如m6A可以直接修飾HK2、GLUT1、ENO1、PDK4等關鍵糖酵解酶的mRNA,通過影響這些酶的表達來調節腫瘤糖酵解。m6A還可以通過調控lncRNA和circRNA的下游通路來調節腫瘤糖酵解。例如OSSC中的circFOXK2/IGF2BP3/GLUT1軸和膠質母細胞瘤中的lncRNA CASC9/IGF2BP2/HK2軸。從目前的研究來看,科學家可以預測m6A通過極其復雜的相互作用網絡調控腫瘤糖酵解,從而影響腫瘤的發生、發展和治療。進一步研究m6A與腫瘤糖酵解之間的潛在聯系將有助于人類更深入地了解腫瘤代謝,并對未來開發新的腫瘤診斷方法和治療方案具有重要意義。
雖然靶向m6a修飾的腫瘤糖酵解治療腫瘤的策略前景廣闊,但相關研究仍存在挑戰。早在20世紀50年代就在生物體中檢測到m6A修飾,但m6A調控腫瘤糖酵解的研究是近年才開始開展的。因此,相關研究尚處于起步階段,還有許多潛在的機制尚未被發現。從代謝的角度來看,腫瘤細胞的異質性使得腫瘤組織中不同的細胞具有不同的代謝狀態,這對靶向糖酵解在癌癥治療中的應用提出了嚴峻的挑戰。目前,臨床前研究已經證明了某些糖酵解抑制劑的抗腫瘤特性,如二甲雙胍和天然化合物白楊素和胡蘿卜素。此外,體外和動物研究表明,抑制結直腸癌細胞中的METTL3可以降低糖酵解水平,恢復對5-FU的治療敏感性。在耐藥乳腺癌細胞中,特定的ALKBH5基因敲除抑制糖酵解并恢復對曲妥珠單抗和拉帕替尼的治療反應。可以預見,m6A抑制劑和腫瘤糖酵解抑制劑的聯合作用將為未來腫瘤的治療打開一扇新的希望之窗。然而,現有的m6A調節劑和腫瘤糖酵解的特異性藥物和小分子抑制劑都處于開發階段,還沒有進入臨床試驗階段。為了盡快應用于臨床,迫切需要尋找抗腫瘤效果強、生物利用度高、藥物副作用小的特異性抑制劑。
綜上所述,葡萄糖代謝與腫瘤增殖、血管生成和淋巴管生成、轉移、免疫逃逸、預后評價和治療密切相關。在不同類型的腫瘤中,m6A修飾通過改變關鍵糖酵解酶mRNA的穩定性,激活或抑制糖酵解相關信號通路,影響腫瘤的生物學行為、治療反應和預后。因此,進一步探索m6A甲基化調控腫瘤糖酵解的復雜關系和機制,為今后的腫瘤治療提供了新的思路。開發針對m6A調節因子和腫瘤糖酵解的特異性抑制劑將是未來腫瘤治療的新方向。
參考文獻
Shi-Wei Yu, Hai-Ling Liu, Hong-Fei S, er al. m6A-regulated tumor glycolysis: new advances in epigenetics and metabolism. Molecular Cancer. 2023(22):137 DOI:10.1186/s12943-023-01841-8