






TNFAIP2通過KEAP1/NRF2信號傳導賦予頭頸部鱗狀細胞癌順鉑耐藥
耐藥限制了順鉑類化療在頭頸部鱗狀細胞癌(HNSCC)中的治療效果,其潛在機制尚不完全清楚。本研究的目的是探討HNSCC順鉑耐藥的原因。在本研究中,TNFAIP2的高表達與HNSCC的不良預后、順鉑耐藥和低活性氧(ROS)水平相關。具體來說,它通過抑制ROS介導的c-JUN N-末端激酶(JNK)磷酸化,保護癌細胞免受順鉑誘導的凋亡。機制上,TNFAIP2中包含的DLG基序通過直接結合KEAP1的Kelch結構域與NRF2競爭,從而阻止NRF2進行泛素蛋白酶體介導的降解。這導致NRF2的積累并產生順鉑耐藥性。在HNSCC標本中證實了TNFAIP2蛋白水平與NRF2及其下游靶基因之間的正相關。我們的研究結果揭示了TNFAIP2/KEAP1/NRF2/ JNK軸在HNSCC中的抗氧化和順鉑耐藥調節作用,提示TNFAIP2可能是改善順鉑治療效果的潛在靶點,特別是對于順鉑耐藥患者。本文于2023年8月發表于“Journal of Experimental & Clinical Cancer Research”(IF=11.3)上。
技術路線

結果
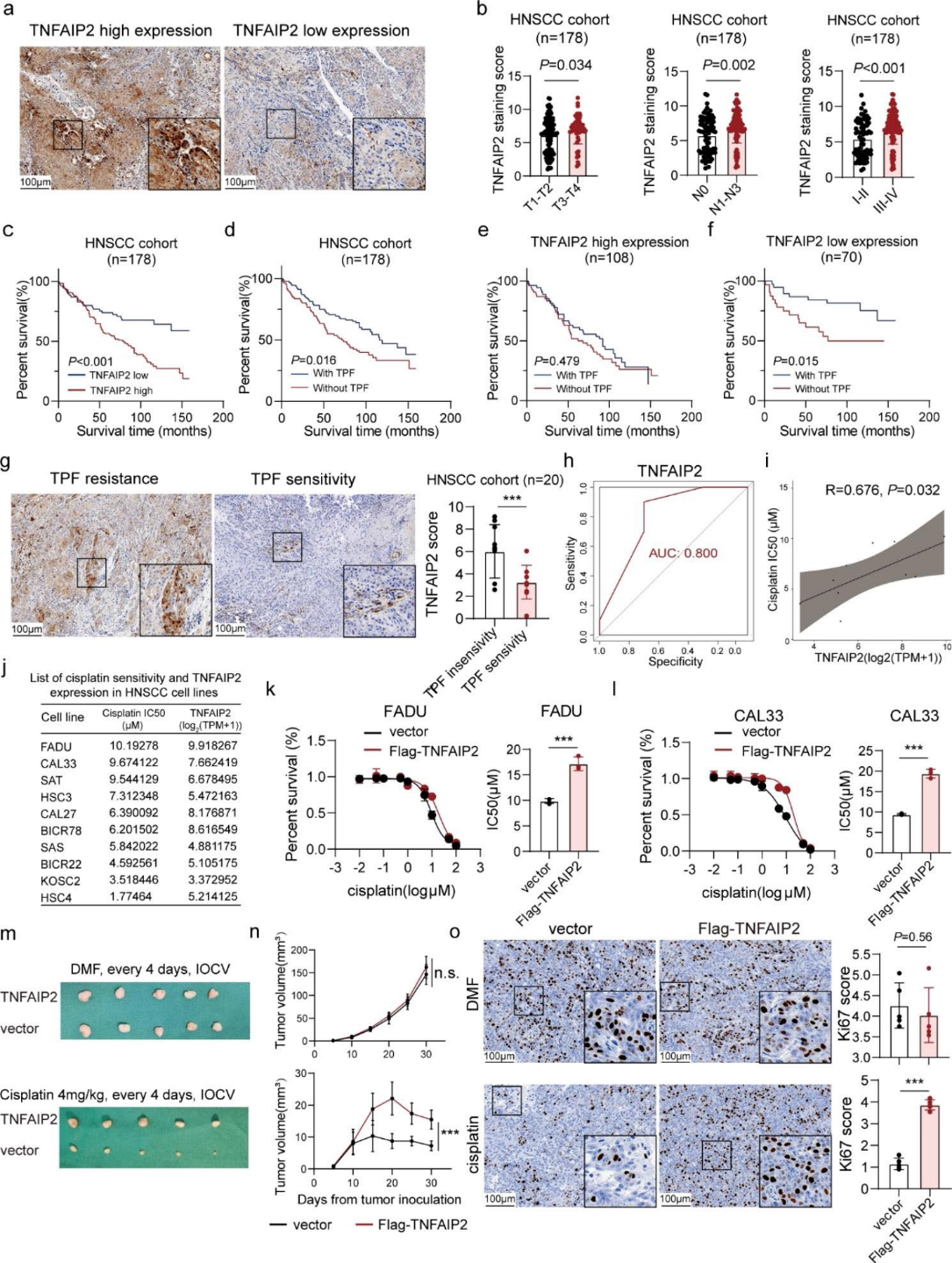
1)TNFAIP2高表達的HNSCC患者易出現順鉑治療失敗
我們對178例HNSCC患者進行了免疫組化染色。我們發現TNFAIP2在晚期患者中表達上調,提示其腫瘤促進作用(圖1a和b)。TNFAIP2高表達表明總生存率較差(圖1c)。我們觀察到TPF化療在TNFAIP2低表達組中取得了更好的治療效果(圖1 -f)。在另一個HNSCC患者單獨接受TPF治療的隊列(n = 20)中,TNFAIP2在TPF治療耐藥患者中顯著上調(圖1g)。表達水平有效地區分了TPF治療的合適候選者(圖1h)。我們進一步研究了10個HNSCC細胞系中TNFAIP2 mRNA水平與紫杉醇、順鉑和5-氟尿嘧啶半IC50之間的相關性。結果顯示,順鉑IC50與TNFAIP2表達呈正相關(圖1i、j)。此外,IC50結果證實,TNFAIP2過表達增加了順鉑耐藥性,而其敲低則降低了順鉑耐藥性(圖1k、l)。隨后,我們通過將FADU皮下接種到BALB/c裸鼠中,并給予指示治療(圖1m),建立了異種移植腫瘤模型。結果顯示,TNFAIP2過表達對腫瘤重量和體積均無影響,但明顯降低了順鉑的治療效果(圖1n)。Ki67染色在體內證實了這種作用(圖1o)。綜上所述,上述結果揭示了TNFAIP2在順鉑治療HNSCC中的不良作用。
2)TNFAIP2通過抑制ROS/JNK信號保護HNSCC細胞免受順鉑誘導的凋亡
根據以往的報道,惡性腫瘤通過多種機制逃避順鉑誘導的細胞凋亡。我們發現當TNFAIP2過表達時,ROS水平降低(圖2a和b)。此外,GSH/GSSG比值以及SOD活性也增加,表明氧化應激減輕(圖2c)。ROS清除劑NAC恢復了TNFAIP2敲低誘導的順鉑耐藥(圖2d-g)。大量證據表明,過量的ROS通過持續激活JNK通路誘導細胞凋亡。我們假設TNFAIP2以ROS/JNK途徑抑制的方式保護癌細胞免受順鉑細胞毒性(圖2h)。結果,JNK抑制劑(SP600125)逆轉了TNFAIP2敲低誘導的細胞凋亡率和順鉑敏感性的增加(圖2i和j)。Western blot分析顯示,當TNFAIP2過表達時,順鉑誘導的JNK磷酸化和順鉑誘導的caspase 9和caspase 3的激活持續被抑制(圖2k)。NAC逆轉TNFAIP2敲低誘導的信號增強(圖21),表明TNFAIP2通過抑制ROS/JNK途徑保護HNSCC細胞免受順鉑誘導的凋亡。

3)TNFAIP2通過抑制NRF2的泛素化和降解來穩定NRF2蛋白
基于TCGA和GEO (GSE39366)數據的進一步富集分析顯示,NRF2信號在TNFAIP2高表達的組織中顯著上調(圖3a)。我們認為TNFAIP2的抗氧化作用是由NRF2介導的。正如預期的那樣,當NRF2被敲除時,TNFAIP2過表達不影響ROS水平(圖3b和c),而促進順鉑耐藥的能力也消失了(圖3d和e)。此外,一組NRF2靶基因,包括HO1、NQO1、GCLC、CAT和SOD2,在mRNA和蛋白水平上與TNFAIP2一起發生改變(圖3f和g)。這些結果表明TNFAIP2的抗氧化和順鉑耐藥功能是由NRF2介導的。然后,我們發現TNFAIP2影響的是NRF2的蛋白水平而不是mRNA水平(圖3 h和i),表明這種調控作用發生在轉錄后階段。經環己亞胺(CHX)處理后,TNFAIP2敲低組NRF2降解更快(圖3j),表明TNFAIP2抑制NRF2蛋白降解,但不促進其合成。隨后,我們發現當TNFAIP2被敲低時,蛋白酶體抑制劑MG132或類泛素化修飾抑制劑MLN4924可阻斷NRF2的降解(圖3k)。此外,免疫共沉淀(Co-IP)實驗顯示,當TNFAIP2被敲除時,泛素結合的NRF2增加,這可以通過TNFAIP2過表達來恢復(圖31)。總之,TNFAIP2通過促進NRF2蛋白穩定表現出抗氧化和順鉑耐藥功能。

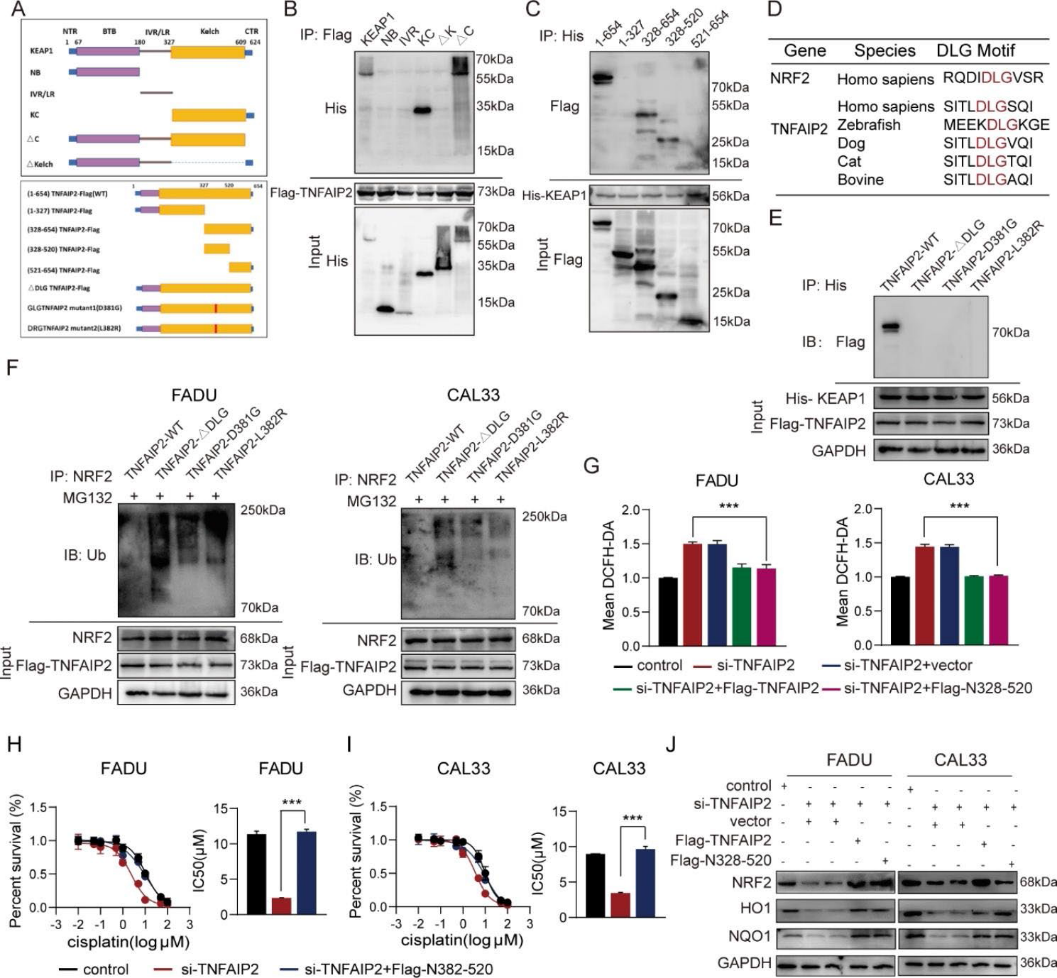
4)TNFAIP2與KEAP1相互作用以穩定NRF2
為了確定TNFAIP2穩定NRF2的機制,我們使用Co-IP/MS進行了高通量篩選。結果顯示TNFAIP2和KEAP1之間存在廣泛的結合關系(圖4a), Co-IP驗證了這一點(圖4b)。我們推測KEAP1可能介導TNFAIP2的氧化應激抑制和順鉑耐藥促進作用。為了驗證這一點,我們敲低了KEAP1,發現TNFAIP2過表達不會導致ROS減少(圖4c)、IC50升高(圖4d和e)、NRF2蛋白積累(圖4f)。Co-IP實驗顯示,當TNFAIP2被敲除時,KEAP1結合了更多的NRF2(圖4g)。相反,當TNFAIP2以濃度梯度過表達時,KEAP1沉淀的NRF2量逐漸減少(圖4h)。

5)DLG基序和kelch結構域介導TNFAIP2與KEAP1的相互作用
我們構建了一系列的截短質粒來探索TNFAIP2和KEAP1之間特異的相互作用位點(圖5a)。首先,將flag標記的TNFAIP2全長和his標記的KEAP1片段質粒共轉染到293T細胞中。co-IP分析顯示,只有含有Kelch結構域的片段才能與TNFAIP2相互作用(圖5b)。同樣,TNFAIP2的全長(N1-654)和其他兩個片段(N328-654和N328-520)也被KEAP1沉淀,這意味著TNFAIP2通過N328-520片段與KEAP1相互作用(圖5c)。有趣的是,我們發現DLG基序恰好位于TNFAIP2的N328-520片段中,該片段在不同物種中高度保守(圖5d)。此外,我們構建了一個DLG缺失(ΔDLG)和兩個突變體flag標記的質粒(圖5a)。正如預期的那樣,三者都不能與KEAP1相互作用(圖5e)。與野生型TNFAIP2組相比,三個質粒轉染組的NRF2泛素化水平均未降低(圖5f)。此外,我們還進行了一系列的拯救實驗來驗證DLG基序的作用。我們發現過表達的N328-520片段逆轉了由TNFAIP2干擾引起的ROS水平升高、順鉑敏感性升高和NRF2豐度降低(圖5g-j)。這些結果表明,DLG基序和Kelch結構域介導了TNFAIP2與KEAP1的相互作用和順鉑耐藥的發展。
6)靶向Tnfaip2可促進體內順鉑治療效果
為了探索抑制TNFAIP2對順鉑治療的促進作用,我們建立了4NQO誘導的HNSCC小鼠模型(圖6a)。在Tnfaip2敲低組中,腫瘤病變面積減少,但當Tnfaip2過表達時,這種效果不顯著。此外,si-Tnfaip2與順鉑聯合使用的腫瘤抑制和細胞凋亡誘導效果最為顯著(圖6b-d)。同時,腫瘤侵襲性明顯受到抑制(圖6e)。GSH/GSSG比值和SOD活性持續下降,表明敲低Tnfaip2會加重腫瘤內氧化應激(圖6f)。免疫組化染色表明當Tnfaip2被抑制時,NRF2及其靶基因在蛋白水平上有效減少(圖6g和h)。以上結果證實了Tnfaip2特異性siRNA可以通過抑制HNSCC中的NRF2信號改善順鉑治療效果。

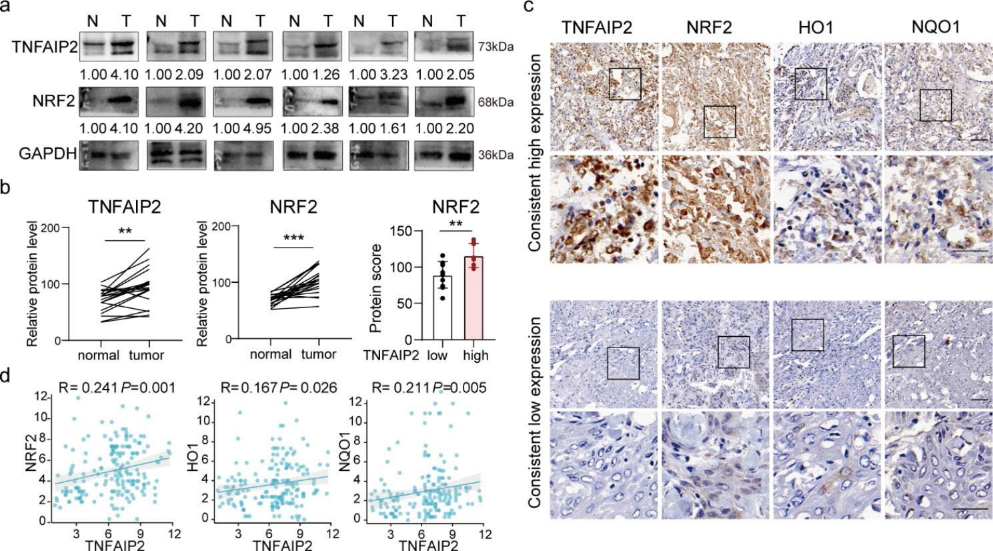
7)HNSCC患者TNFAIP2表達與NRF2表達呈正相關
為了驗證TNFAIP2和NRF2在HNSCC中的表達水平及其相關性,我們首先使用western blotting檢測了20對HNSCC組織中TNFAIP2和NRF2的表達。與鄰近正常組織相比,TNFAIP2和NRF2在HNSCC組織中的表達更高(圖7a)。在TNFAIP2低表達的組織中,NRF2也下調(圖7 b)。免疫組化染色顯示TNFAIP2與NRF2、HO1和NQO1呈正相關(圖7c和d),這表明TNFAIP2可能在HNSCC中作為NRF2信號啟動子。
結論
TNFAIP2可以通過與KEAP1的競爭性相互作用來穩定NRF2,從而抑制順鉑誘導的氧化應激和細胞凋亡,最終導致順鉑耐藥。TNFAIP2可能是改善HNSCC化療治療效果的潛在治療靶點。
實驗方法
WB,Co-IP/MS,RT?qPCR,CCK-8,克隆形成實驗,流式,ROS和抗氧化能力檢測,免疫組化,TUNEL染色,雙熒光素酶報告基因實驗。
參考文獻
Xu T, Yang Y, Chen Z, Wang J, Wang X, Zheng Y, Wang C, Wang Y, Zhu Z, Ding X, Zhou J, Li G, Zhang H, Zhang W, Wu Y, Song X. TNFAIP2 confers cisplatin resistance in head and neck squamous cell carcinoma via KEAP1/NRF2 signaling. J Exp Clin Cancer Res. 2023 Aug 1;42(1):190. doi: 10.1186/s13046-023-02775-1.