






心肌梗死后,乳酸誘導(dǎo)Snail1乳酸化促進(jìn)內(nèi)皮-間充質(zhì)轉(zhuǎn)化

高乳酸水平與心臟病患者的預(yù)后和死亡率呈正相關(guān)。內(nèi)皮-間質(zhì)轉(zhuǎn)化(EndoMT)在心臟纖維化中起著重要作用。本研究中,作者報(bào)告了乳酸的新功能,即在心肌梗死(MI)后,通過(guò)促進(jìn)EndoMT增加心臟纖維化并加劇心功能不全。機(jī)制上,乳酸能上調(diào)缺氧/ MI后Snail1的核轉(zhuǎn)位和乳酸化。抑制Snail1可改善MI后乳酸誘導(dǎo)的心臟功能障礙,抑制乳酸刺激的EndoMT、Snail1的乳酸化和TGF-β/Smad2信號(hào)的激活。該研究于2023年2月發(fā)表在《Science Advances》,IF:13.6。
技術(shù)路線





主要研究結(jié)果
1. 減少乳酸改善MI后的心功能和心臟纖維化
腹腔注射糖酵解抑制劑2-脫氧-D-葡萄糖(2-DG)降低乳酸水平。2-DG 抑制MI引起的血清和心臟乳酸水平(圖1A、B)。值得注意的是,2-DG MI 小鼠的射血分?jǐn)?shù)(EF%)和縮短分?jǐn)?shù)(FS%)水平高于對(duì)照組MI小鼠(圖1C、D)。2-DG MI小鼠的左心室舒張末期容積(LVEDV)和左心室收縮末期容積(LVESV)值低于對(duì)照MI小鼠(圖E、F)這表明抑制乳酸生成改善MI后的心臟功能。此外,Masson三色染色顯示給予2-DG減少M(fèi)I后的心臟纖維化(圖 1G)。這些數(shù)據(jù)表明乳酸的產(chǎn)生促進(jìn)MI誘發(fā)的纖維化和心功能不全。
2. 增加乳酸生成加劇MI后的心功能不全和心臟纖維化
在誘導(dǎo)MI3小時(shí)后給小鼠補(bǔ)充乳酸,在MI1周和4周后用超聲心動(dòng)圖評(píng)估心臟功能。圖1H顯示,腹腔注射乳酸(0.5 g/kg體重7天后,血清乳酸濃度恢復(fù)到正常水平。與假手術(shù)組相比,MI 顯著降EF% 和FS% 的值(圖1I 、J)與 MI 組相比,注射乳酸進(jìn)一步降低EF%(7 天時(shí)降低14.0%,28 天時(shí)降低19.5%)和FS%(7 天時(shí)降低15.8%,28 天時(shí)降低21.6%)的值(圖1I、J)。同樣,乳酸處理也增加MI后LVEDV和LVESV的水平(圖1K、L)這些數(shù)據(jù)共同表明,乳酸水平升高加重MI后的心臟功能障礙。Masson三色染色表明乳酸給藥顯著誘導(dǎo)MI小鼠的心臟纖維化(圖1M)。

圖1. 乳酸水平升高導(dǎo)致MI后心臟功能障礙惡化和心臟纖維化增加
3. 抑制乳酸生成減輕MI后的心臟EndoMT
與假手術(shù)組相比,MI誘導(dǎo)內(nèi)皮標(biāo)志物CD31和VE-cadherin(VE-鈣粘蛋白)表達(dá)(圖 2A)以及間充質(zhì)標(biāo)志物成纖維細(xì)胞特異性蛋白1(FSP1)、α-平滑肌肌動(dòng)蛋白(α-SMA)和Collagen1a1 表達(dá)(圖 2A)明顯增加。施用2-DG抑制乳酸產(chǎn)生提高 CD31和VE-cadherin的表達(dá),同時(shí)消除MI誘導(dǎo)的 Collagen1a1、α-SMA和FSP1 的減少(圖2A)。同樣,用2-DG 處理上調(diào)內(nèi)皮標(biāo)志物Cdh5和Kdr的mRNA水平(圖2B)。相反,2-DG下調(diào)間質(zhì)標(biāo)志物Atca2、Col1a1、Fn1和S100a4的mRNA 表達(dá)(圖2B)。總之,這些數(shù)據(jù)表明抑制乳酸減弱MI后的EndoMT進(jìn)程。在添加或不添加2-DG的內(nèi)皮細(xì)胞特異性綠色熒光蛋白(GFP)標(biāo)記小鼠(TIE2GFP)中誘導(dǎo)MI,并用抗GF(綠色)和抗FSP1(紅色)抗體對(duì)心臟組織進(jìn)行免疫熒光染色。如圖2C所示,與假手術(shù)組相比,MI 誘導(dǎo)GFP與FSP1的共定位。與此相反,2-DG抑制乳酸產(chǎn)生減輕MI誘導(dǎo)的GFP與FSP1的共定位,表明2-DG減少M(fèi)I刺激的心肌EndoMT。此外,在MI或假手術(shù)后7天,從獲得的心臟中分離出內(nèi)皮細(xì)胞,流式細(xì)胞術(shù)檢測(cè)分化成肌成纖維細(xì)胞的內(nèi)皮細(xì)胞百分比。如圖2D所示,從假手術(shù)組小鼠體內(nèi)分離的內(nèi)皮細(xì)胞顯示,抗 CD31 和抗 CD114(內(nèi)皮細(xì)胞標(biāo)記物)抗體染色的GFP細(xì)胞最多,幾乎檢測(cè)不到Gp38(心臟成纖維細(xì)胞標(biāo)記物)染色(圖2E)。然而,MI明顯增加GFP標(biāo)記的內(nèi)皮細(xì)胞中Gp38陽(yáng)性染色細(xì)胞的數(shù)量和百分比。相反,用2-DG處理可抑制MI 誘導(dǎo)的Gp38陽(yáng)性內(nèi)皮細(xì)胞。
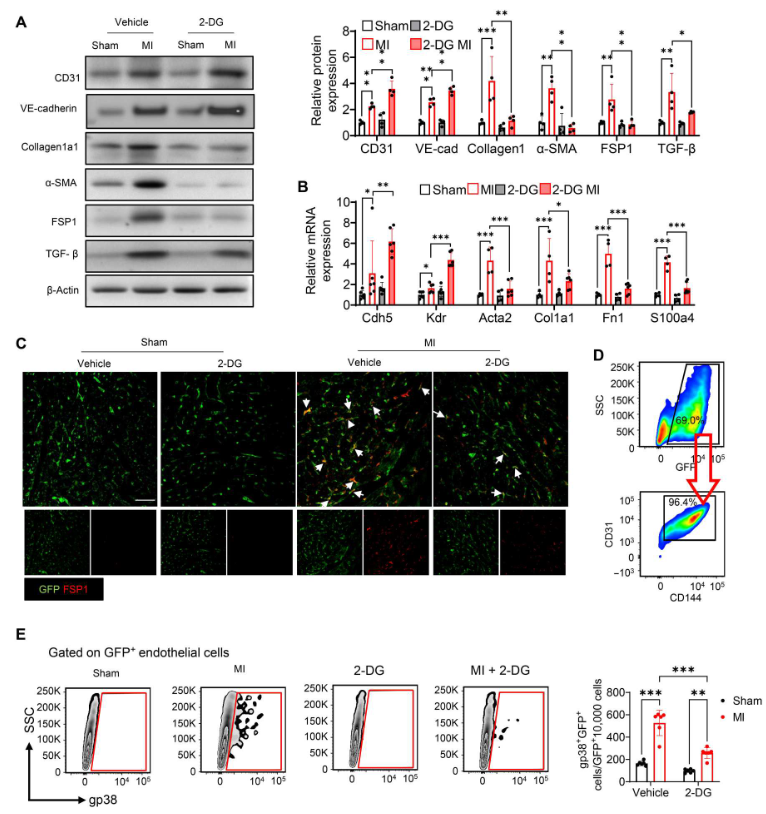
圖2. 2-DG減弱MI誘導(dǎo)的EndoMT
4. 高水平乳酸促進(jìn)MI后心肌中的EndoMT
WB顯示,對(duì)MI小鼠補(bǔ)充乳酸降低內(nèi)皮細(xì)胞標(biāo)記物(CD31和VE-cadherin)的水平,促進(jìn)間充質(zhì)標(biāo)記物(FSP1、α-SMA和Collagen1a1)的表達(dá)(圖3A)qRT-PCR顯示,乳酸水平升高導(dǎo)致MI后Cdh5和Kdr mRNA水平降低(圖3B、C),并誘導(dǎo)Col1a1、Fn1和S100a4 mRNA水平升高(圖3D- F),表明乳酸誘導(dǎo)MI 后的EndoMT。用抗 GFP(綠色)和抗 FSP1(紅色)抗體進(jìn)行免疫熒光染色顯示,與假手術(shù)組小鼠相比,MI 誘導(dǎo)GFP和FSP1的共定位(圖3G),這表明乳酸鹽處理后有更多的內(nèi)皮細(xì)胞分化成成纖維細(xì)胞。同樣,流式細(xì)胞術(shù)顯示,MI 后乳酸鹽處理進(jìn)一步上調(diào)GFP+gp38+ 細(xì)胞的數(shù)量(圖3H),表明乳酸鹽促進(jìn)MI后的EndoMT。

圖3. 補(bǔ)充乳酸鹽促進(jìn)MI后的EndoMT
5. 缺氧后,乳酸鹽刺激內(nèi)皮細(xì)胞遷移、降低VE-cadherin的表達(dá),并促EndoMT
進(jìn)行傷口愈合試驗(yàn)和EdU摻入試驗(yàn),圖4A顯示,與常氧對(duì)照組相比,缺氧加速內(nèi)皮細(xì)胞的遷移。此外,10 mM(而非5 mM)的乳酸顯著促進(jìn)缺氧誘導(dǎo)的傷口愈合(圖4A),表明乳酸鹽能誘導(dǎo)缺氧挑戰(zhàn)下的內(nèi)皮細(xì)胞遷移。同樣,EdU摻入試驗(yàn)再次證明乳酸鹽能促進(jìn)缺氧刺激的內(nèi)皮細(xì)胞增殖(圖4B)。VE-cadherin的免疫熒光染色顯示,缺氧72小時(shí)后,VE-cadherin的完整性受到破壞(圖4D)。缺氧后,用10 mM乳酸鹽處理進(jìn)一步破壞內(nèi)皮細(xì)胞表面的VE-cadherin完整性(圖4D)。此外,WB和 qRT-PCR 分析顯示,與缺氧組相比,乳酸處理顯著降低VE-cadherin的水平(圖4C、5C),表明乳酸改變?nèi)毖跆魬?zhàn)下內(nèi)皮細(xì)胞的表型。用乳酸處理內(nèi)皮細(xì)胞,檢測(cè)缺氧后EndoMT的標(biāo)記物。如圖5A所示,缺氧72小時(shí)后,內(nèi)皮細(xì)胞的形態(tài)從典型形狀變?yōu)槔L(zhǎng)的紡錘形外觀。與缺氧對(duì)照組相比,乳酸處理導(dǎo)致細(xì)胞形態(tài)發(fā)生更大變化。此外,拉長(zhǎng)細(xì)胞的FSP1染色呈陽(yáng)性,CD31 染色呈陰性(圖5B)。圖5C顯示,缺氧挑戰(zhàn)后,乳酸減少VE-cadherin的表達(dá)并誘導(dǎo)α-SMA的釋放。進(jìn)行基于Matrigel的內(nèi)皮細(xì)胞血管生成試驗(yàn),發(fā)現(xiàn)乳酸的施用抑制缺氧誘導(dǎo)的血管生成(圖5D)。此外,利用膠原凝膠收縮試驗(yàn),證實(shí)乳酸處理的內(nèi)皮細(xì)胞在缺氧刺激后表現(xiàn)出類似間充質(zhì)的功能,這表現(xiàn)在膠原凝膠體積減小和收縮力增強(qiáng)(圖5E)。總之,這些數(shù)據(jù)表明缺氧后乳酸誘導(dǎo)EndoMT。
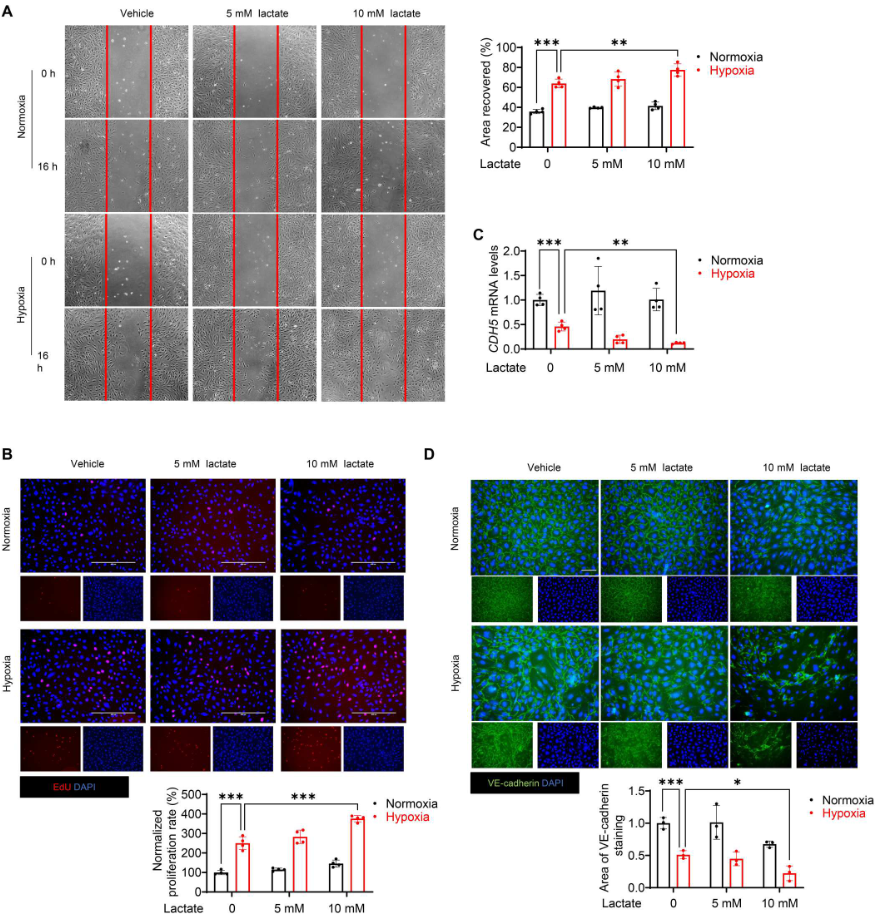
圖4. 缺氧后,乳酸誘導(dǎo)內(nèi)皮細(xì)胞遷移并減少VE-cadherin的表達(dá)
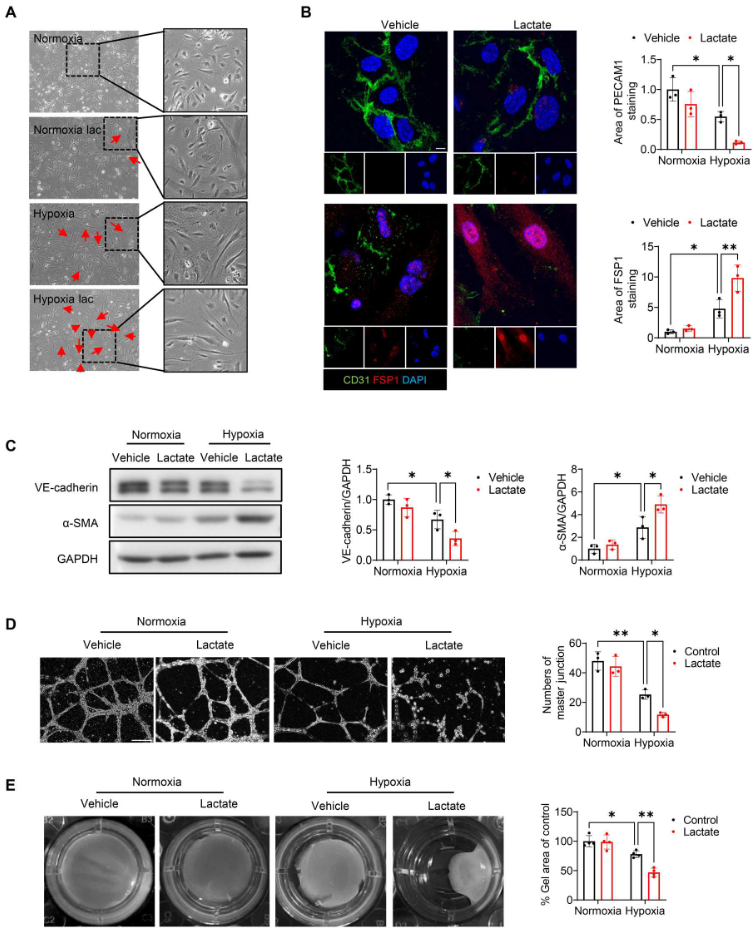
圖5. 缺氧后,乳酸促進(jìn)內(nèi)皮細(xì)胞中EndoMT
6. 缺氧后乳酸激活TGF-β/Smad2信號(hào)傳導(dǎo)
qRT-PCR顯示,在體內(nèi)乳酸顯著加速M(fèi)I后心肌中Tgfb1和Smad2 mRNA的表達(dá)(圖6A、B)。然而,乳酸鹽并沒(méi)有改變Smad3的mRNA水平(圖6C)。此外,2-DG抑制乳酸的產(chǎn)生降低MI誘導(dǎo)的Tgfb1和Smad2 mRNA表達(dá)(圖6D、E)。體外研究也表明,乳酸促進(jìn)缺氧挑戰(zhàn)下的人臍帶內(nèi)皮細(xì)胞(HUVECs)和HCMECs中TGFB1和SMAD2 mRNA 的表達(dá)(圖6F、G)。與基因表達(dá)水平一致,乳酸處理上調(diào)缺氧挑戰(zhàn)后Smad2的磷酸化和TGF-β的表達(dá),但不影響 Smad3的磷酸化(圖6H)。這些數(shù)據(jù)綜合表明,乳酸誘導(dǎo)的TGF-β/Smad2激活有助于缺氧后的EndoMT。
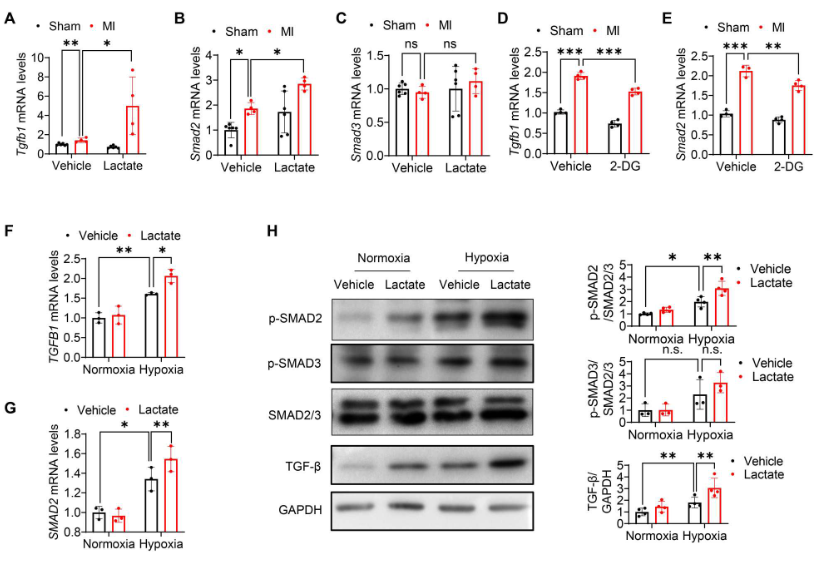
圖6. MI/缺氧后,乳酸刺激TGF-β/Smad2信號(hào)傳導(dǎo)
7. 抑制細(xì)胞內(nèi)乳酸減輕缺氧誘導(dǎo)的EndoMT
CHC[單羧酸鹽轉(zhuǎn)運(yùn)體(MCT)抑制劑α-氰基-4-羥基肉桂酸鹽]降低乳酸處理誘導(dǎo)的細(xì)胞內(nèi)乳酸水平(圖7A),表明細(xì)胞外乳酸是通過(guò)MCT 轉(zhuǎn)運(yùn)到內(nèi)皮細(xì)胞的。此外,服用CHC改善缺氧后乳酸促進(jìn)的內(nèi)皮細(xì)胞遷移(圖7B)。免疫熒光染色顯示,CHC阻乳酸處理導(dǎo)致的VE-cadherin表達(dá)減少(圖7C),而血管形成試驗(yàn)表明,CHC促進(jìn)缺氧后的血管生成(圖7D)。此外,CHC還緩解乳酸誘導(dǎo)的內(nèi)皮標(biāo)志物PECAM1、CDH5和KDR mRNA水平的降低(圖7E-G)以及間充質(zhì)標(biāo)志物ACTA2、FN1和COL1A1 mRNA水平的升高(圖7H-J)。用CHC處理抑制乳酸誘導(dǎo)的TGFB1和SMAD2 mRNA的表達(dá)(圖7K、L)。
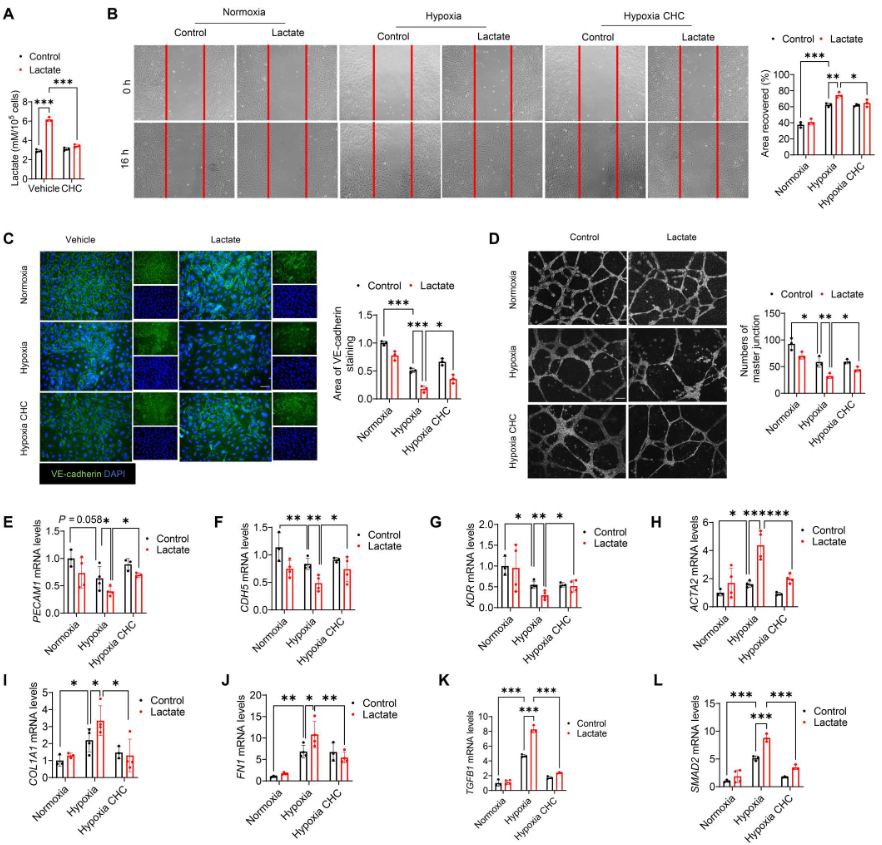
圖7. 抑制細(xì)胞內(nèi)乳酸減弱缺氧誘導(dǎo)的EndoMT
8. 缺氧后乳酸促進(jìn)Snail1的核轉(zhuǎn)位
如圖8A所示,單獨(dú)缺氧不會(huì)改變Snail1的胞漿表達(dá)。然而,與常氧狀態(tài)相比,缺氧后Snail1的核表達(dá)明顯加快。乳酸處理進(jìn)一步上調(diào)Snail1的核轉(zhuǎn)位。免疫熒光染色顯示,缺氧后Snail1核表達(dá)的陽(yáng)性率更高,而乳酸進(jìn)一步誘導(dǎo)Snail1核表達(dá)(圖8B),表明Snail1的核轉(zhuǎn)位在乳酸促進(jìn)的EndoMT中發(fā)揮重要作用。用抗Snail1抗體進(jìn)行染色質(zhì)免疫沉淀(ChIP)實(shí)驗(yàn),圖8C顯示,在缺氧挑戰(zhàn)的細(xì)胞中,Snail1與TGFB1 基因一起被大量招募。然而在乳酸處理的缺氧挑戰(zhàn)細(xì)胞中,TGFB1基因和Snail1蛋白之間的相互作用明顯大于缺氧細(xì)胞(圖8C)。這些數(shù)據(jù)表明,乳酸促進(jìn)Snail1蛋白和TGFB1基因的相互作用,從而有助于TGF-β/Smad2介導(dǎo)的EndoMT。
9. 缺氧后乳酸誘導(dǎo)Snail1乳酸化
使用抗Snail1抗體進(jìn)行免疫沉淀,然后使用抗泛乙酰賴氨酸(Kac)或抗泛乳酸賴氨酸(Kla)抗體進(jìn)行免疫印跡,結(jié)果顯示缺氧誘導(dǎo)Snail1的乙酰化和乳酸化。給予乳酸進(jìn)一步上調(diào)Snail1的乙酰化和乳酸化(圖8D)。Snail1和CBP/p300之間存在相互作用,缺氧后乳酸處理促進(jìn)它們之間的相互作用(圖8D)。然而,給予CHC抑制細(xì)胞內(nèi)乳酸可減輕乳酸促進(jìn)的Snail1和CBP/p300之間的相互作用,以及Snail1的乙酰化和乳酸化(圖8E)。反過(guò)來(lái),與缺氧組相比,乳酸處理也顯著增加抗CBP抗體、抗p300抗體、抗pan-Kla抗體和抗pan-Kac抗體免疫沉淀中Snail1的表達(dá)(圖8F)。相比之下,給予CHC可減少Snail1與CBP/p300 之間的相互作用以及Snail1的乳酸化和乙酰化(圖8F)。
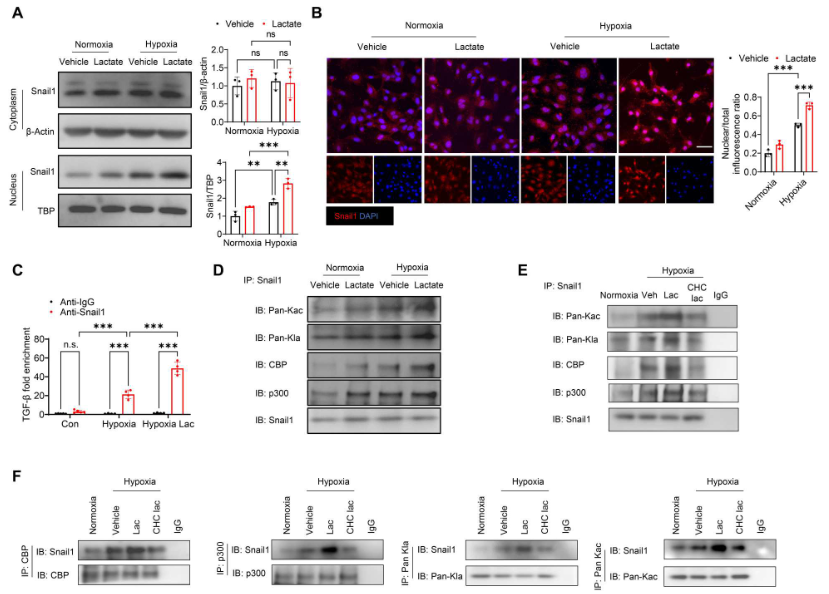
圖8. 缺氧后,乳酸促進(jìn)Snail1的核轉(zhuǎn)位和乳酸化
10. 沉默Snail1減弱缺氧后的EndoMT和TGF-β/Smad2激活
通過(guò)轉(zhuǎn)染特異性siRNA沉默Snail1逆轉(zhuǎn)缺氧后乳酸誘導(dǎo)的CD31和VE-cadherin下調(diào)(圖9A)。相反,抑制Snail1阻止乳酸促進(jìn)的間充質(zhì)標(biāo)志物α-SMA 和FSP1的表達(dá)(圖9A)、SMAD2磷酸化和TGF-β激活(圖9H)。qRT-PCR觀察到在乳酸處理下,敲除Snail1提高PECAM1、KDR和CDH5的mRNA水平(圖9B-D),降低ACTA2、COL1A1和S100A4的mRNA水平(圖9E-G)。此外,沉默Snail1降低乳酸誘導(dǎo)的SMAD2和TGFB1 mRNA水平(圖9I、J)。
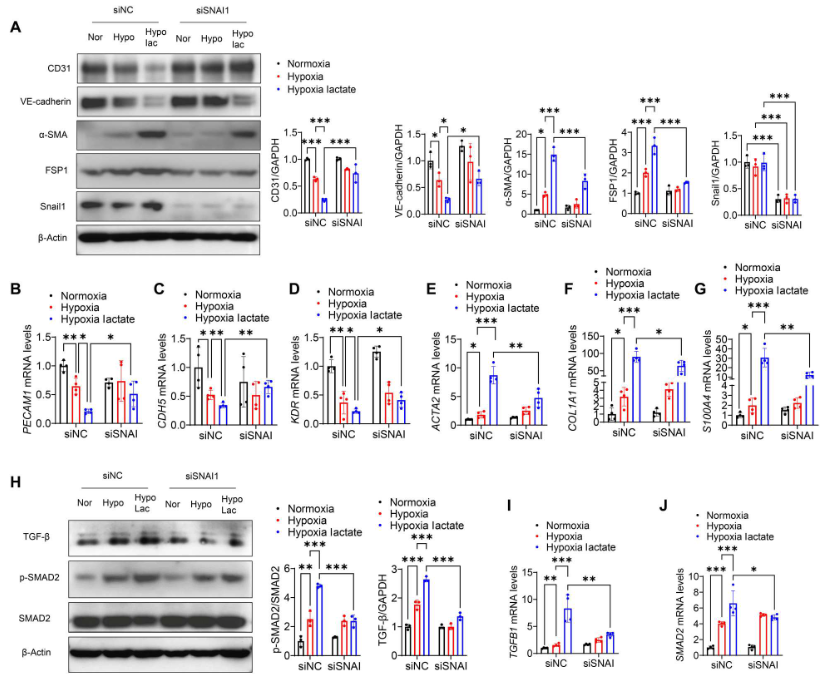
圖9. 沉默Snail1減輕缺氧后的EndoMT和TGF-β/Smad2的激活
11. 沉默Snail1改善MI后的心臟功能并減弱EndoMT
如圖10(A和B)所示,與假手術(shù)組相比,MI顯著促進(jìn)Snail1的乳酸化和乙酰化,以及Snail1與CBP/p300之間的相互作用。乳酸給藥進(jìn)一步誘導(dǎo)它們之間的相互作用(圖10A)。用2-DG處理減輕MI誘導(dǎo)的Snail1與CBP或p300之間的相互作用,并減少Snail1乙酰化和乳酸化(圖10B)。轉(zhuǎn)染特異性Snail1 siRNA,WB和免疫熒光染色證實(shí),施用siSnai1顯著降低Snail1在心臟中的表達(dá)(圖10C、D)。然而,沉默Snail1逆轉(zhuǎn)乳酸降低的EF%和FS%水平(圖10E、F),并防止MI后乳酸誘導(dǎo)的LVEDV和LVESV水平升高(圖10G、H),表明Snail1的缺失改善乳酸誘導(dǎo)的心臟功能障礙。此外,與MI后乳酸處理相比,沉默Snail1提高Cdh5和Kdr mRNA的表達(dá)(圖10I、J),降低Acta2、Fn1和S100a4 mRNA的表達(dá)(圖10K-M),表明Snail1有助于MI后乳酸誘導(dǎo)的EndoMT。這些數(shù)據(jù)表明,Snail1通過(guò)激活TGF-β/Smad2信號(hào)在乳酸促進(jìn)的EndoMT中發(fā)揮重要作用(圖10N)。
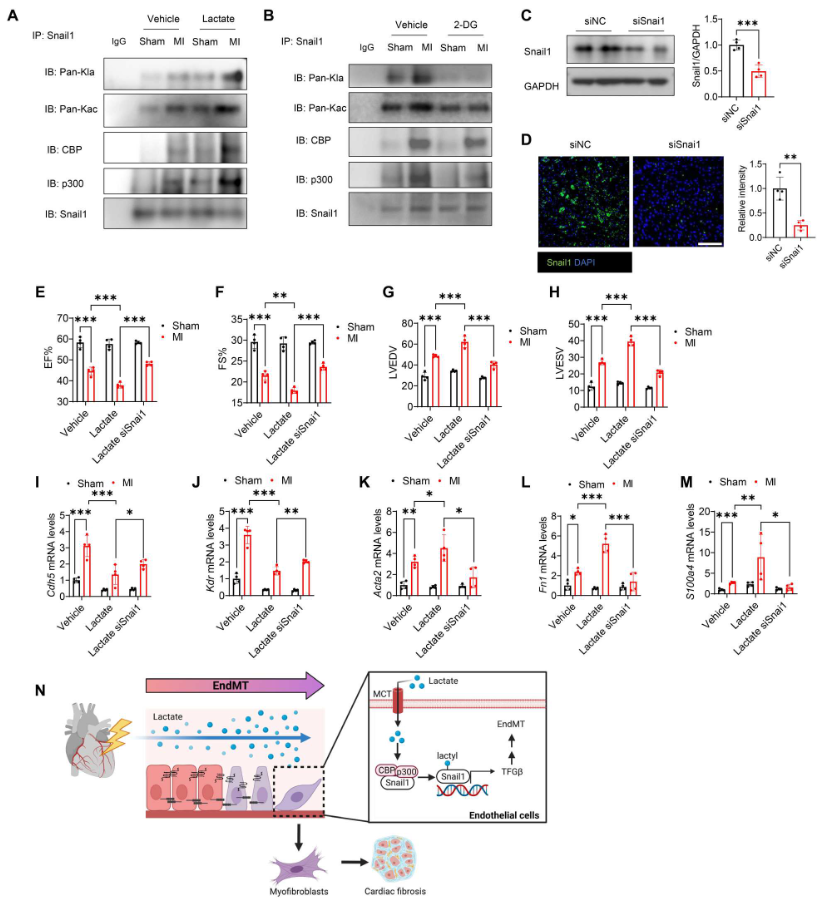
圖10. 沉默Snail1減輕MI后Snail1的乳酸化和EndoMT
結(jié)論
綜上所述,本研究發(fā)現(xiàn)乳酸發(fā)揮一種以前未知的功能,即在MI及缺氧后,乳酸通過(guò)激活TGF-β/ Smad2通路誘導(dǎo)EndoMT增加心臟纖維化并加劇心功能不全。這些發(fā)現(xiàn)加深了我們對(duì)乳酸在MI后EndoMT中作用的理解,并將成為開(kāi)發(fā)創(chuàng)新療法以改善MI后心臟重塑和功能的基礎(chǔ)。
實(shí)驗(yàn)方法
siRNA體內(nèi)轉(zhuǎn)染,Masson三色染色,2,3,5-三苯基氯化四氮唑染色,2,3,5-三苯基氯細(xì)胞凋亡檢測(cè),乳酸測(cè)定,流式細(xì)胞術(shù),小鼠心臟內(nèi)皮細(xì)胞的分離,細(xì)胞培養(yǎng),增殖測(cè)定,血管生成試驗(yàn),膠原凝膠收縮實(shí)驗(yàn),免疫熒光,WB,qRT-PCR,免疫沉淀,ChIP-qPCR
參考文獻(xiàn)
Fan M, Yang K, Wang X, Chen L, Gill PS, Ha T, Liu L, Lewis NH, Williams DL, Li C. Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci Adv. 2023 Feb 3; 9 (5): eadc9465. doi: 10.1126/ sciadv. adc9465. Epub 2023 Feb 3. PMID: 36735787; PMCID: PMC9897666.