






Kremen2通過阻止SOCS3介導的EGFR降解來驅動非小細胞肺癌的進展

肺癌是一種常見的惡性腫瘤,是全球癌癥相關死亡率的主要原因。非小細胞肺癌(NSCLC)約占肺癌的85%。NSCLC患者臨床預后較差,總體5年生存率為15%。靶向治療的出現使NSCLC的治療取得了顯著進展,然而患者不可避免地會出現耐藥。因此,需要基于生物標志物的新靶點。Kremen2是一種由473個氨基酸組成的單跨膜結構域蛋白,是經典Wnt信號通路的重要調節因子。有研究報道,根據癌癥基因組圖譜(TCGA)分析,在來自18種不同癌癥類型的70%以上組織樣本中,腫瘤組織中的Kremen2表達水平顯著高于配對的正常組織,尤其是在Kremen2表達水平比高10倍以上的鱗狀肺癌中。然而,其在NSCLC中的生物學功能和臨床意義尚不明確。表皮生長因子受體(EGFR)是一種受體酪氨酸激酶,是目前公認的治療NSCLC的有效靶點。EGFR的異常活化被認為是惡性轉化和腫瘤轉移的主要原因。多種機制可誘導EGFR的致癌激活,如基因突變、轉錄過表達和EGFR降解缺陷。多項研究表明,EGFR的穩定性是調節肺癌進展的重要決定因素,EGFR降解失調加速了腫瘤的發生和進展。SOCS蛋白家族是眾所周知的細胞因子受體信號轉導的負調控因子。SOCS3是SOCS蛋白家族的成員,通過抑制JAK/STAT信號通路發揮腫瘤抑制作用。敲除SOCS3會破壞E3連接酶復合物的形成,并驅動整合素β1介導的小細胞肺癌轉移。在之前的研究中,SOCS3主要是基于其甲基化沉默導致功能喪失而在NSCLC中進行評估。該研究發表在《Journal of Experimental & Clinical Cancer Research》,IF:11.3。
技術路線

主要研究結果
1. Kremen2在非小細胞肺癌中表達上調,并與不良預后相關
為了尋找新的與NSCLC相關的致癌基因,作者在TCGA數據庫中檢索了504例有病理信息的肺腺癌樣本,并篩選出57對配對的非癌和癌樣本。進一步分析這些配對患者樣本的RNA-seq結果,并使用一般線性模型(P<0.05)篩選差異表達基因(DEGs)(圖1A)。分析顯示,Kremen2的表達存在顯著差異,這在肺癌中尚未被研究過。此外,比較每對TCGA樣本的原始Kremen2 RNA-seq數據后,腫瘤組織中的Kremen2表達水平較高(圖1B)。采用GEPIA軟件分析Kremen2在NSCLC中的表達。結果顯示,與癌旁正常肺組織相比,在NSCLC組織中,尤其是肺鱗狀細胞癌組織中,Kremen2表達顯著上調(圖1C)。
為了評估Kremen2在NSCLC臨床標本中的表達,作者檢測了新鮮冷凍的NSCLC標本及其配對的正常組織中Kremen2 mRNA的水平。結果顯示,與正常組織相比,NSCLC組織中Kremen2 mRNA水平顯著升高(圖1D)。蛋白質印跡法檢測到大多數NSCLC標本中Kremen2蛋白水平升高(圖1E)。免疫組織化學染色進一步顯示,腫瘤組織中Kremen2高表達(圖1F)。此外,作者檢測了一些NSCLC細胞系中的Kremen2蛋白水平,發現在NSCLC細胞中Kremen2蛋白表達普遍較高(圖1G)。
為了闡明Kremen2過表達與NSCLC患者生存的相關性,作者使用Kaplan-Meier plotter數據庫分析NSCLC病例。結果如圖1H和I所示,表明高Kremen2表達水平的NSCLC患者與較差的總生存期(OS)相關,如較低的OS和首次進展生存期(P<0.05)。卡方檢驗顯示,Kremen2表達上調與原發腫瘤(T)分期(P=0.041)和病理分級(P=0.045)相關。綜上所述,這些數據表明,Kremen2在NSCLC中過表達,并且Kremen2過表達可能與患者的不良預后相關。
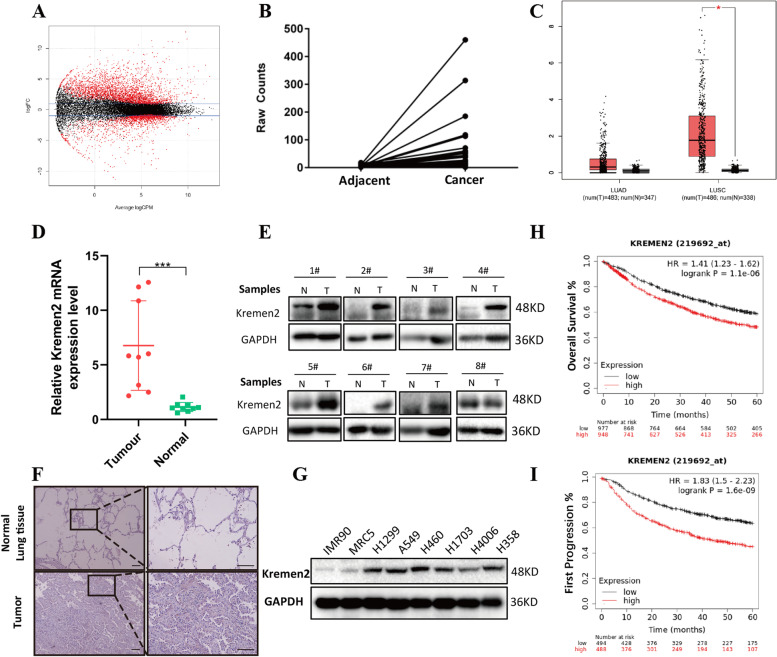
圖1 Kremen2在非小細胞肺癌中表達上調,并與不良預后相關
2. 沉默NSCLC細胞中Kremen2表達可抑制細胞增殖
為了研究Kremen2在NSCLC中的生物學功能,作者利用CRISPR/Cas9方法產生敲除Kremen2的A549細胞(A549-krm2ko),其中插入胸腺嘧啶殘基導致移碼突變。通過克隆形成實驗和EdU摻入實驗檢測Kremen2對A549和A549-krm2ko細胞增殖的影響。A549-Krm2KO細胞的克隆形成能力低于野生型細胞(圖2A),EdU陽性細胞顯著減少,表明細胞增殖能力下降,而在A549-Krm2KO細胞中過表達外源性Flag-Kremen2(Flag-Krm2)時,edu陽性細胞比例增加(圖2B),證實了Kremen2對細胞增殖的影響。此外,作者利用慢病毒介導的shRNA沉默內源性Kremen2表達,建立了穩定沉默Kremen2的NSCLC細胞株。同樣,A549和H1703細胞(shKrm2)中的Kremen2敲低抑制了集落形成和細胞增殖能力(圖2D-F)。通過皮下注射穩定敲低Kremen2的A549細胞建立裸鼠移植瘤模型,驗證這些體外發現是否與體內腫瘤生長相關。在A549異種移植模型中獲得了一致的結果,敲低Kremen2的表達顯著抑制了腫瘤的大小和重量(圖2G-I)。IHC染色顯示,由于A549細胞的Kremen2敲低,A549種植小鼠組織中的ki67陽性細胞百分比較低(圖2J)。綜上所述,這些數據表明,Kremen2在NSCLC細胞生長中發揮重要作用。

圖2 沉默NSCLC細胞中內源性Kremen2表達可抑制細胞增殖
3. Kremen2高表達促進NSCLC細胞轉移
臨床資料顯示,Kremen2的表達與LUAD患者病理分級呈正相關,提示Kremen2可能參與了腫瘤的轉移。為了解決這一問題,作者檢測了體內外Kremen2對腫瘤細胞轉移能力的影響。Transwell實驗結果顯示,在A549細胞中,敲除Kremen2可減弱細胞的侵襲能力,而在A549-krm2ko細胞中過表達Flag-Krm2可增強細胞的侵襲能力(圖3A)。同樣,通過shRNA下調Kremen2表達后,H1703和A549細胞的侵襲數量減少(圖3B,C)。此外,來自傷口愈合實驗的數據表明,敲低Kremen2降低了細胞遷移(圖3D-G)。通過實時熒光定量PCR檢測沉默Kremen2后NSCLC細胞中上皮-間充質轉化(EMT)標志物的變化,探討其對NSCLC細胞EMT的抑制作用。Kremen2敲低和低pro-EMT Snail1和Twist1水平之間存在明顯的正相關。為了進一步了解Kremen2在體內是否促進腫瘤轉移,作者對A549-Krm2KO細胞和A549-Krm2KO + OE-Krm2細胞(A549-Krm2KO細胞過表達Kremen2)進行了尾靜脈轉移實驗。結果顯示,刪除Kremen2減少了肺轉移結節的數量,而過表達Kremen2顯著增加了肺轉移結節的數量(圖3H-J)。為了更好地觀察肺內轉移結節的形成,作者在A549細胞中構建了共表達GFP的shCtrl和shKrm2細胞,觀察細胞的熒光強度(圖3K和L)。將兩種表達GFP的細胞注射到裸鼠尾靜脈。HE染色和熒光信號結果顯示,在A549細胞中下調Kremen2與對照組相比,肺轉移結節數量減少(圖3M-O)。綜上所述,這些發現表明,Kremen2是NSCLC轉移的促進因子。
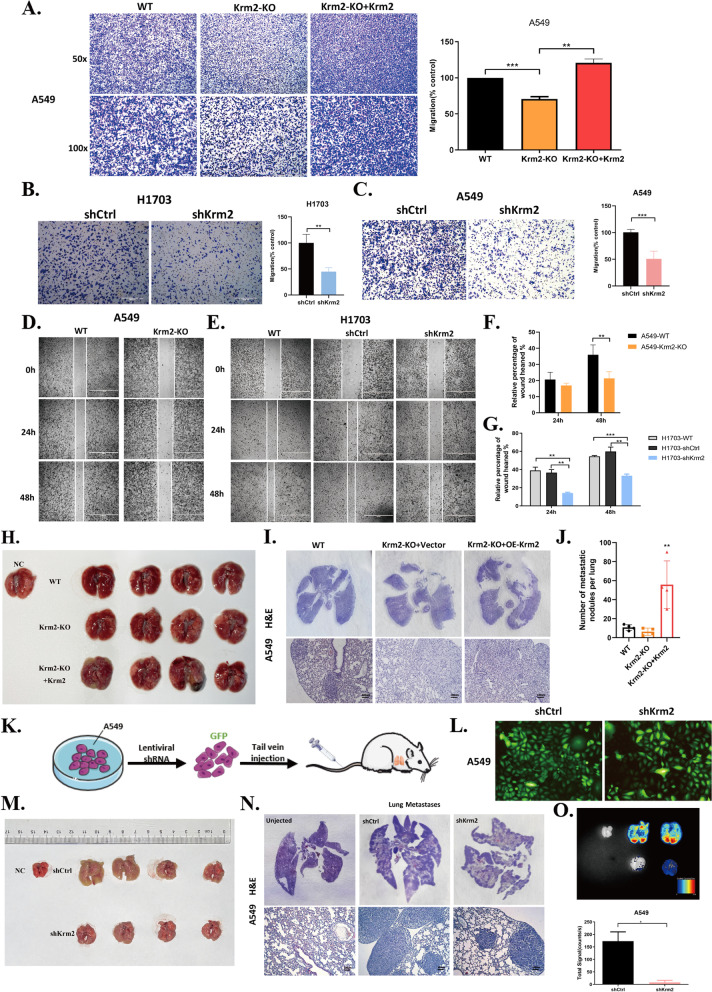
圖3 體內外實驗顯示,Kremen2促進NSCLC細胞轉移
4. Kremen2參與多種癌癥相關信號通路
為了闡明Kremen2促進肺癌發生的機制,采用RNA-seq比較H1703細胞中對照組和Kremen2敲低(shKrm2)組的轉錄組變化。根據表達的倍數變化(≥1或≤-1),shKrm2組中共鑒定出273個上調基因和281個下調基因。對上調和下調基因進行層次聚類分析(圖4A)。GO分析表明,DEGs與細胞增殖、發育、運動和遷移密切相關(圖4B)。KEGG富集分析表明,這些DEGs富集在與癌癥相關的信號通路,如MAPK、Wnt和PI3K-AKT信號通路(圖4C)。由于之前的Kremen2研究多集中在Wnt信號通路,作者首先檢測到了Wnt信號通路中關鍵蛋白表達的變化。蛋白質印跡結果顯示,敲低Kremen2并沒有顯著影響β-catenin蛋白水平,但增加了H1703細胞中LRP6的總蛋白和磷酸化水平(圖4D),可能是由于LRP6的內吞降解減少。此外,在H1703細胞中敲低Kremen2后,PI3K(Y607)和AKT(Ser473)的磷酸化水平顯著下調(圖4E),這與在胃癌中對Kremen2的研究結果一致。
由于STAT3信號通路的激活已經在相當比例的NSCLC病例中被報道,作者也測試了Kremen2是否調節STAT3信號通路。蛋白質印跡結果顯示磷酸化JAK2(Y1007和Y1008)和STAT3(Y705)水平下調,而總JAK2和STAT3水平無變化。與此同時,STAT3通路中負責調節細胞增殖的下游因子c-Myc和Cyclin D1蛋白水平也顯著降低(圖4F和G)。相反,過表達Kremen2增強了p-JAK2、p-STAT3、c-Myc和Cyclin D1蛋白水平(圖4H),表明Kremen2影響了JAK2-STAT3信號通路。

圖4 抑制Kremen2參與多種癌癥相關信號通路
5. Kremen2通過穩定EGFR促進NSCLC細胞增殖
考慮到EGFR是JAK2-STAT3和PI3K-AKT信號通路的上游共同激活因子,在NSCLC的進展中發揮重要作用,作者將從shKrm2 H1703細胞中篩選的DEGs輸入FunRich軟件進行功能分析。結果顯示大多數基因聚集在EGFR相關通路上。然后,作者檢測了Kremen2是否影響EGFR蛋白水平。蛋白質印跡結果表明,siRNA介導沉默Kremen2可下調A549和H1703細胞中的EGFR蛋白水平(圖5A)。在Kremen2穩定敲低的細胞中,EGFR蛋白水平也降低,而過表達Kremen2增加了EGFR蛋白水平(圖5B)。作者在A549-Krm2KO細胞中獲得了類似的結果。將Flag-tagged Kremen2重新引入A549-Krm2KO細胞中可恢復EGFR蛋白水平(圖5C)。如圖5D所示,蛋白酶體抑制劑MG132逆轉了Kremen2敲除細胞中EGFR蛋白水平的下降,表明Kremen2可能通過蛋白酶體降解來調節EGFR蛋白水平。作者進一步研究了敲低Kremen2是否影響EGF刺激后EGFR的磷酸化。結果顯示,沉默Kremen2降低了A549和H1703細胞中EGF誘導的EGFR、STAT3和AKT的磷酸化(圖5E)。為了評估Kremen2是否通過調節EGFR促進細胞增殖,作者在過表達Kremen2的A549細胞中去除內源性EGFR,觀察到抑制內源性EGFR的表達減弱了Kremen2誘導的細胞增殖(圖5F)。相反,在Kremen2敲低的A549細胞中過表達EGFR表明外源性EGFR表達促進了Kremen2敲低細胞的增殖(圖5H)。綜上所述,這些數據表明,在NSCLC中,Kremen2的高表達水平可能通過維持EGFR的穩定性促進腫瘤進展。
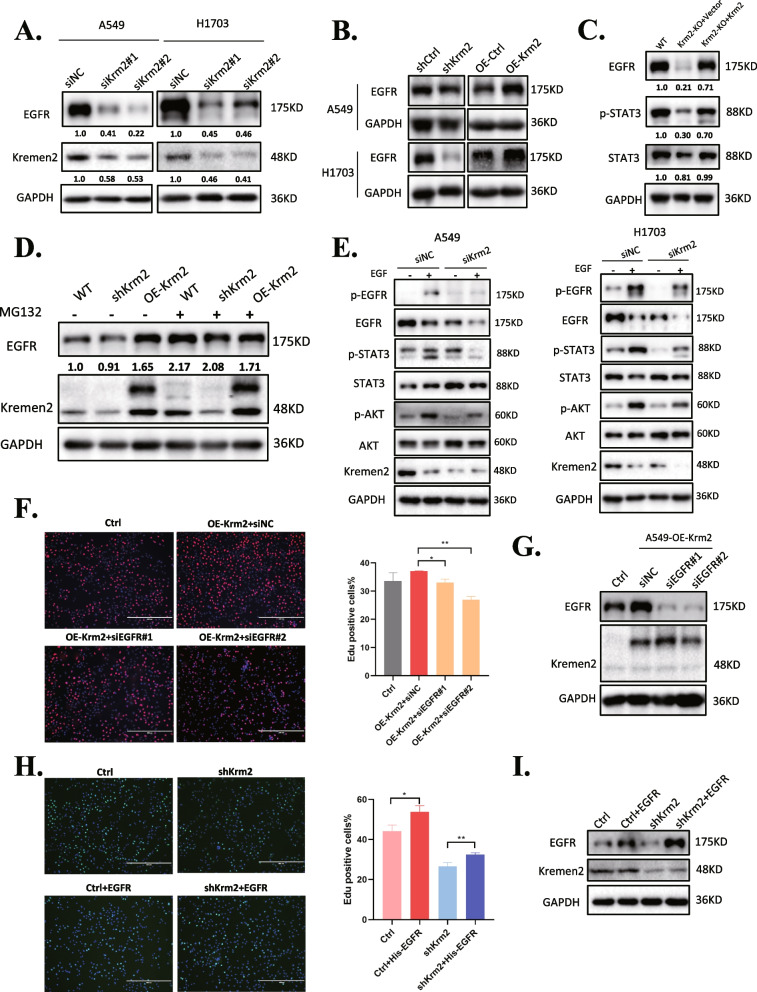
圖5 Kremen2通過促進EGFR的穩定促進NSCLC細胞增殖
6. Kremen2和SOCS3的相互作用可抑制EGFR的泛素化和降解
為了了解Kremen2是如何調控EGFR的,作者使用co-IP來下調參與EGFR信號通路的Kremen2相互作用蛋白。令人驚訝的是,Kremen2不與EGFR、AKT或STAT3蛋白相互作用,但與STAT3信號通路的負調控因子SOCS3強相關(圖6A)。此外,在A549細胞(圖6B)中觀察到內源性Kremen2和SOCS3共定位。在變性條件下純化外源性SOCS3,然后對Kremen2進行免疫印跡分析,證實了Kremen2和SOCS3之間的相互作用(圖6C)。接下來,作者分別在A549和A549-oe-krm2細胞中過表達SOCS3,以探索SOCS3在Kremen2和EGFR之間關系中的作用。如圖6D所示,過表達外源性SOCS3降低了A549細胞中EGFR蛋白水平和STAT3的磷酸化水平,而穩定過表達Kremen2部分逆轉了這一現象(圖6D)。
此外,SOCS3過表達細胞中EGFR水平的下降被蛋白酶體抑制劑MG132逆轉,表明SOCS3通過蛋白酶體降解調節EGFR水平(圖6E)。為了進一步研究SOCS3是如何調控EGFR的,作者用cicloheximide處理細胞并檢測EGFR的半衰期。結果表明,過表達SOCS3可縮短EGFR蛋白的半衰期(圖6F)。接下來,作者對內源性蛋白進行了相互共IP。正如預期的那樣,內源性SOCS3與EGFR相互作用(圖6G),而內源性EGFR與SOCS3結合(圖6H)。這些結果表明SOCS3可以與EGFR結合并促進細胞中EGFR的降解。
據報道,SOCS3可促進TBK1和整合素β1的泛素化和降解;因此,作者接下來研究了SOCS3對EGFR泛素化的影響。結果,與對照組相比,在A549細胞中過表達SOCS3增加了EGFR的多泛素化(圖6I)。為了進一步研究Kremen2是否影響SOCS3介導的EGFR泛素化,作者進行了體外泛素化實驗。在HEK293T細胞中,Flag-SOCS3和His-EGFR共表達,在變性條件下純化His-EGFR,隨后對EGFR進行免疫印跡分析,證明在SOCS3過表達細胞中外源性EGFR泛素化增加,而Kremen2過表達導致SOCS3對EGFR的多泛素化減少(圖6J)。
綜上所述,這些數據表明,Kremen2與SOCS3相互作用,并通過阻止SOCS3介導的EGFR泛素化和降解來穩定EGFR蛋白;從而增強EGFR信號通路的活化,促進NSCLC細胞的增殖和轉移。

圖6 Kremen2與SOCS3的相互作用抑制EGFR的泛素化和降解
結論
該研究證明了Kremen2在NSCLC腫瘤發生中的新作用。機制上,Kremen2可能通過與SOCS3相互作用,阻斷泛素依賴的EGFR降解,從而維持EGFR介導的腫瘤信號通路,促進NSCLC細胞的增殖和轉移。因此,Kremen2可能成為NSCLC新的治療靶點。
機制圖

實驗方法
免疫組織化學(IHC),菌落形成和EdU測定,流式細胞術,Transwell和傷口愈合測定,定量實時聚合酶鏈反應(qRT-PCR),蛋白質印跡,免疫沉淀,免疫熒光,動物實驗
參考文獻
Sun Y, Gao Y, Dong M, Li J, Li X, He N, et al. Kremen2 drives the progression of non-small cell lung cancer by preventing SOCS3-mediated degradation of EGFR. J Exp Clin Cancer Res. 2023 Jun 3;42(1):140.