






新的鐵死亡調(diào)控回路——蛋白激酶ATM通過磷酸化協(xié)調(diào)鐵自噬

鐵死亡是一種由細(xì)胞內(nèi)生物活性鐵催化的脂質(zhì)過氧化物的致死積累驅(qū)動(dòng)的程序性細(xì)胞死亡。Ser/Thr蛋白激酶ATM,DNA雙鏈斷裂損傷的主要傳感器,是鐵死亡執(zhí)行不可或缺的。本研究中,作者發(fā)現(xiàn)ATM激酶支配細(xì)胞內(nèi)不穩(wěn)定的鐵池,或者通過磷酸化鐵自噬受體NCOA4,從而操縱鐵自噬的鐵周轉(zhuǎn)。研究結(jié)果揭示了一種新的調(diào)控回路,包括ATM-NCOA 4在協(xié)調(diào)鐵自噬和鐵生物利用度。該研究于2023年6月發(fā)表于《Autophagy》上,IF=13.3。
技術(shù)路線









主要研究?jī)?nèi)容
1.ATM激酶在細(xì)胞鐵死亡中的作用
為了證實(shí)ATM激酶的鐵結(jié)合調(diào)節(jié)能力,使用ATM敲除MEF細(xì)胞。我們發(fā)現(xiàn)ATM敲除保護(hù)MEF免于由erastin和RSL 3觸發(fā)的細(xì)胞死亡,如細(xì)胞活力測(cè)定所證明的(圖1A和1B)。補(bǔ)充erastin或RSL 3導(dǎo)致WT細(xì)胞中明顯的PI陽性染色,其幾乎可以被Fer完全阻斷。ATM敲除顯著拮抗這種PI陽性染色,如熒光顯微鏡掃描和流式細(xì)胞術(shù)分析所示(圖1C-1F)。此外,erastin和RSL 3的暴露誘導(dǎo)脂質(zhì)過氧化物的過度產(chǎn)生,其在ATM敲除MEF細(xì)胞中被顯著抑制(圖1G和1H)。總的來說,這些數(shù)據(jù)證實(shí)了ATM激酶在鐵蛋白減少過程中不可或缺的作用。此外,我們還證實(shí)了ATM激酶在其他誘導(dǎo)劑誘導(dǎo)的鐵死亡中的相關(guān)性。丁硫氨酸亞砜亞胺(BSO)和柳氮磺胺吡啶(Sul)分別通過不可逆地抑制γ-谷氨酰半胱氨酸合酶(γ-glutamylcysteine synthase,γ-GS)和強(qiáng)有力地靶向系統(tǒng)Xc-,消耗細(xì)胞內(nèi)GSH,從而啟動(dòng)鐵代謝。ATM缺失也抵消了BSO和硫誘導(dǎo)的鐵死亡,如細(xì)胞活力測(cè)定(圖1I和1J)、PI染色(圖1K-1N)和脂質(zhì)過氧化分析(圖1O和1P)所示。總之,這些發(fā)現(xiàn)表明ATM激酶對(duì)于鐵磷酸化執(zhí)行是不可或缺的。

圖1:ATM激酶在細(xì)胞鐵死亡中是不可或缺的
2.ATM決定的鐵死亡不依賴于TRP53
腫瘤抑制因子TRP53是ATM激酶最重要的下游靶點(diǎn)之一。更重要的是,已報(bào)道TRP 53通過多種機(jī)制操縱鐵凋亡,而不僅僅是其對(duì)細(xì)胞凋亡和自噬的充分表征的調(diào)節(jié)作用。為了研究TRP53在ATM介導(dǎo)的鐵凋亡中的潛在作用,分析了在TRP53消融的遺傳背景下ATM敲除MEFs和對(duì)照MEFs的鐵凋亡敏感性。在本文中,AP29細(xì)胞是TRP53單敲除的,而AP26細(xì)胞是ATM TRP53雙敲除的(圖2A)。與WT MEF相比,TRP53單敲除AP29細(xì)胞在一定程度上對(duì)erastin和RSL 3誘導(dǎo)的細(xì)胞死亡更具抗性(圖2B和2C),表明TRP53的促鐵蛋白激酶活性。當(dāng)與TRP53單敲除AP29細(xì)胞相比時(shí),ATM TRP53雙敲除AP26細(xì)胞似乎對(duì)erastin和RSL3誘導(dǎo)的鐵死亡更加不敏感,如細(xì)胞活力測(cè)定(圖2B和2C)、PI染色(圖2D)和脂質(zhì)過氧化分析(圖2 E和2F)所示。值得注意的是,在暴露于erastin和RSL3的AP26細(xì)胞中觀察到Ptgs2的表達(dá)降低(圖2G和2 H)。為了進(jìn)一步證實(shí)TRP53在ATM介導(dǎo)的鐵磷貯積癥中的相關(guān)性,使用了匹氟菊酯-α(PFTα),一種抑制TRP 53響應(yīng)基因的TRP53依賴性反式激活的特異性TRP 53抑制劑。補(bǔ)充PFTα可明顯下調(diào)Cdkn1a/p21(細(xì)胞周期蛋白依賴性激酶抑制劑1A(P21);一種重要的TRP53靶向基因)和MEF細(xì)胞中Slc7a11的上調(diào)(圖2 I),表明PFTα確實(shí)抑制TRP53的轉(zhuǎn)錄因子活性。在PFTα攻擊期間,在ATM KO細(xì)胞中觀察到Cdknla的邊際下調(diào)和Slc7al1的上調(diào)(圖2 I),這可能是由于ATM KO細(xì)胞中TRP53的較低表達(dá)(圖2A)。值得注意的是,PFTα補(bǔ)充在WT和ATM KO細(xì)胞中均驅(qū)動(dòng)鐵凋亡抗性(圖2 J、2K),進(jìn)一步表明TRP53的鐵凋亡促進(jìn)能力。另外,在WT和ATM KO細(xì)胞中,更長(zhǎng)時(shí)間的erastin或RSL 3處理導(dǎo)致更多的細(xì)胞死亡。具體地,當(dāng)與PFTα處理的WT細(xì)胞相比時(shí),PFTα處理的ATM KO細(xì)胞對(duì)erastin和RSL3表現(xiàn)出高得多的不敏感性(圖2 J、2K)。總之,這些研究表明ATM決定的細(xì)胞鐵死亡,至少部分獨(dú)立于TRP53。
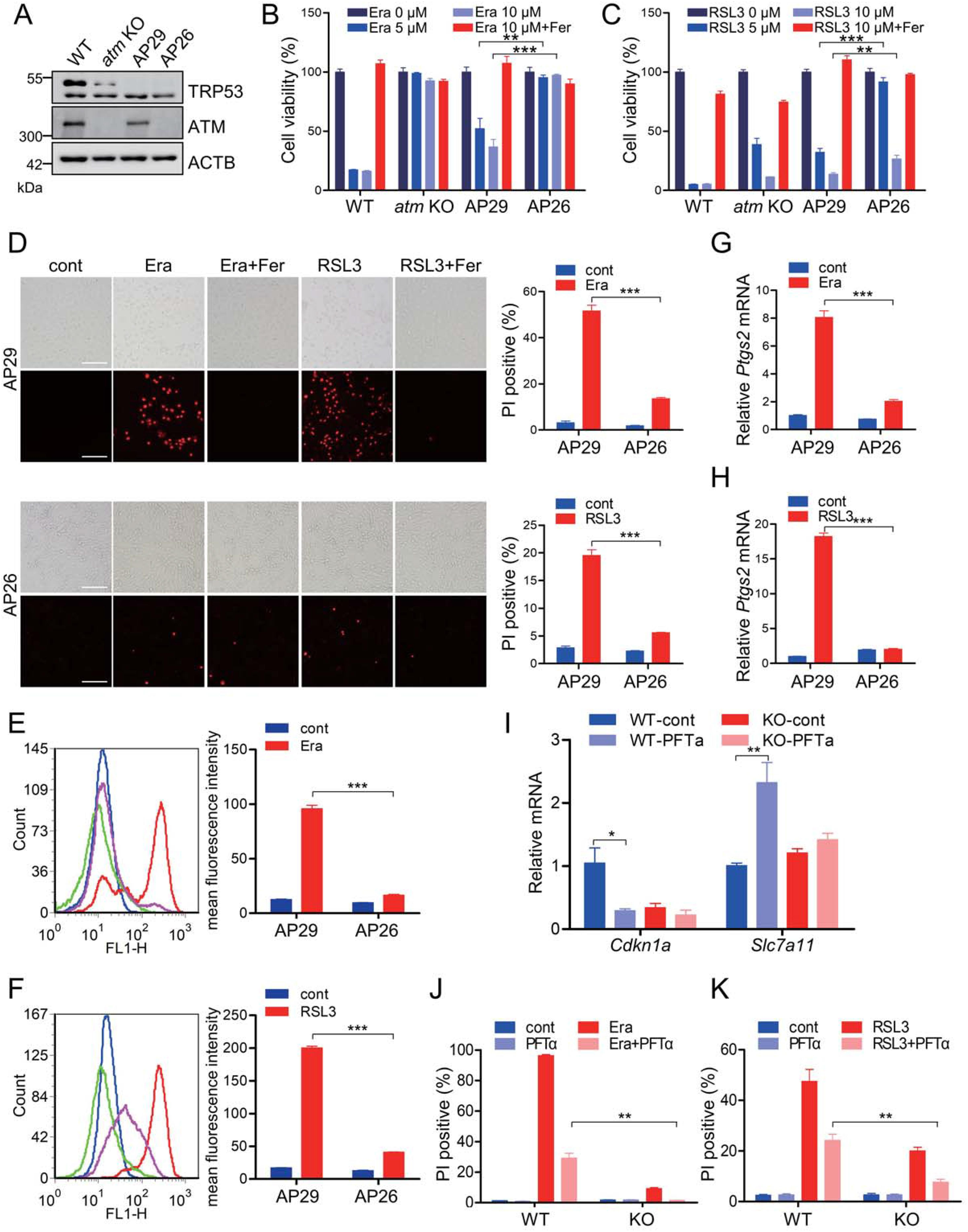
圖2:ATM決定的鐵死亡不依賴于TRP53
3.ATM決定了與鐵蛋白相關(guān)的鐵自噬
細(xì)胞內(nèi)不穩(wěn)定的游離鐵是決定氧化還原穩(wěn)態(tài)的重要因素之一。細(xì)胞可滲透的FerroOrange是一種新型的熒光鐵傳感器,其能夠?qū)崿F(xiàn)細(xì)胞內(nèi)亞鐵的活細(xì)胞熒光成像。鐵凋亡誘導(dǎo)劑導(dǎo)致WT MEF細(xì)胞中FerroOrange的熒光增強(qiáng),而在相同處理下在ATM KO MEF細(xì)胞中該熒光大大減弱(圖3A-3C)。此外,在具有穩(wěn)定ATM敲低的HT-1080中也觀察到類似的表型(圖3D-3F),表明ATM在鐵死亡執(zhí)行期間支配細(xì)胞內(nèi)不穩(wěn)定游離鐵的觀點(diǎn)。然而,鐵外排輸出蛋白SLC40A1/FPN(溶質(zhì)載體家族40成員1)和鐵儲(chǔ)存相細(xì)胞溶質(zhì)鐵主要被鐵蛋白納米籠隔離。據(jù)報(bào)道,NCOA4介導(dǎo)的鐵自噬在細(xì)胞鐵死亡誘導(dǎo)期間被激活,而一般自噬組分或特異性鐵自噬受體NCOA4的耗竭急劇降低鐵動(dòng)員,導(dǎo)致細(xì)胞不穩(wěn)定鐵庫減少并拮抗鐵細(xì)胞凋亡。erastin暴露導(dǎo)致WT MEF細(xì)胞中FTHl蛋白增加(圖3G)。在WT MEF細(xì)胞中,在erastin暴露期間還觀察到mRNA水平上的升高的FTHl表達(dá),如定量RTPCR測(cè)定所示(圖3I)。FTH1在ATM KO MEF細(xì)胞穩(wěn)定狀態(tài)下高表達(dá)。然而,在erastin暴露期間,在ATM KO細(xì)胞中未觀察到FTHl蛋白或mRNA的明顯增加,表明在ATM消融期間鐵蛋白周轉(zhuǎn)減慢(圖3G和3I)。另外,這在具有RSL3處理的ATM KO細(xì)胞中是類似的(圖3 H)。為了進(jìn)一步證實(shí)ATM在確定鐵凋亡相關(guān)的鐵蛋白周轉(zhuǎn)中的重要作用,利用ATM敲低HT-1080細(xì)胞,并且我們發(fā)現(xiàn)當(dāng)ATM沉默時(shí),F(xiàn)THl也高度表達(dá)(圖3 J和3 K)。類似地,erastin和RSL3暴露導(dǎo)致對(duì)照細(xì)胞中FTHl的增加。ATM敲低導(dǎo)致在erastin和RSL3暴露下HT-1080細(xì)胞中的惰性FTHl升高(圖3 J和3 K)。此外,erastin暴露導(dǎo)致FTHl轉(zhuǎn)錄的明顯增加,這在ATM敲低HT-1080中未觀察到(圖3L)。免疫熒光染色顯示,erastin處理促進(jìn)鐵蛋白和溶酶體的共定位,而這種共定位在ATM KO MEF細(xì)胞和ATM敲低HT-1080細(xì)胞中受到明顯抑制(圖3M-3 P)。總之,這些數(shù)據(jù)表明,ATM在鐵蛋白周轉(zhuǎn)中主導(dǎo)了與鐵死亡相關(guān)的鐵自噬。
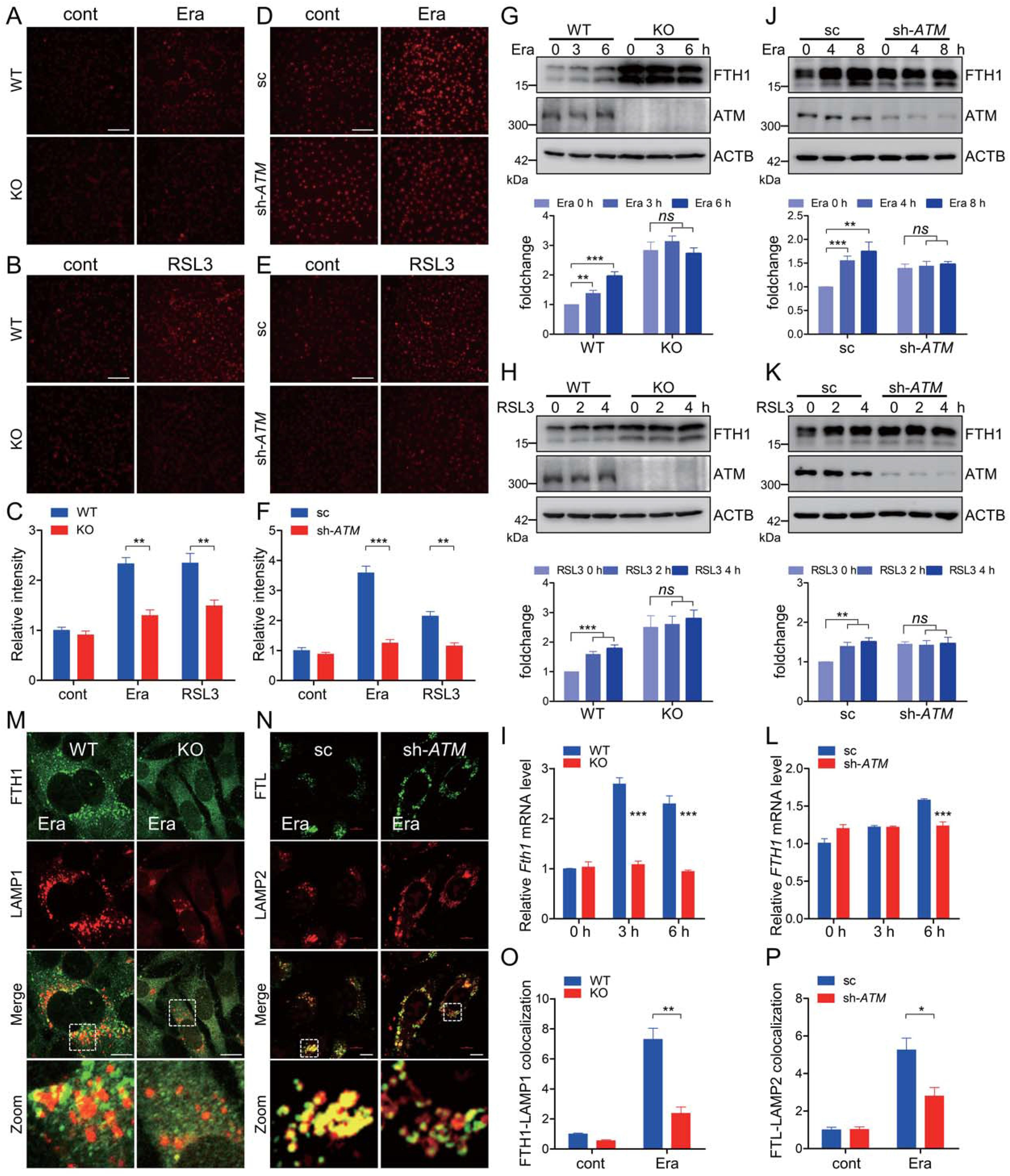
圖3:ATM決定了與鐵蛋白相關(guān)的鐵自噬
4.ATM主導(dǎo)鐵饑餓誘導(dǎo)的鐵自噬
ATM是否僅操縱特異性鐵蛋白沉積相關(guān)的鐵自噬或ATM是否能在遺傳上調(diào)節(jié)鐵饑餓誘導(dǎo)的鐵自噬是下一個(gè)重要的問題。暴露的鐵螯合劑,包括去鐵酮(DFP)和去鐵胺(DFO),導(dǎo)致FTH1在MEF細(xì)胞中的快速降解。ATM敲除減弱了DFP和DFO誘導(dǎo)的FTH1降解(圖4A和4B)。另外,ATM抑制劑KU 55933的預(yù)處理也阻斷了這種FTHl降解(圖4C和4D)。此外,鐵螯合劑促進(jìn)FTH1和溶酶體標(biāo)志物L(fēng)AMP1的共定位,以及鐵自噬受體NCOA4和自體吞噬體標(biāo)志物GFP-MAP1LC3的共定ATM的異位表達(dá)加速了DFO誘導(dǎo)的FTHl降解(圖4G)。自噬抑制劑氯喹(CQ)急劇地阻止了這種DFO誘導(dǎo)的FTHl降解,即使在ATM過表達(dá)的細(xì)胞中也是如此(圖4G)。研究發(fā)現(xiàn),ATG9敲除和通過其抑制劑PIK-III 的PIK3C3/VPS34抑制可以在對(duì)照細(xì)胞和ATM過度表達(dá)細(xì)胞中干擾鐵螯合劑誘導(dǎo)的FTHl降解(圖4 H-4J)。這些數(shù)據(jù)進(jìn)一步證實(shí)了ATM激酶在鐵饑餓誘導(dǎo)的鐵自噬中的調(diào)節(jié)作用。ATM操縱鐵饑餓誘導(dǎo)的鐵蛋白吞噬似乎是TRP53非依賴性的,因?yàn)楫?dāng)與TRP53單敲除AP29細(xì)胞相比時(shí),在ATM TRP53雙敲除AP26細(xì)胞中仍然觀察到減慢的FTHl降解(圖4K和4L)。總之,這些發(fā)現(xiàn)表明ATM是鐵自噬的主要調(diào)節(jié)劑。然而,在ATM抑制期間,這兩種共定位均被顯著抑制(圖4 E和4F)。總之,這些發(fā)現(xiàn)表明ATM是鐵自噬的主要調(diào)節(jié)因子。
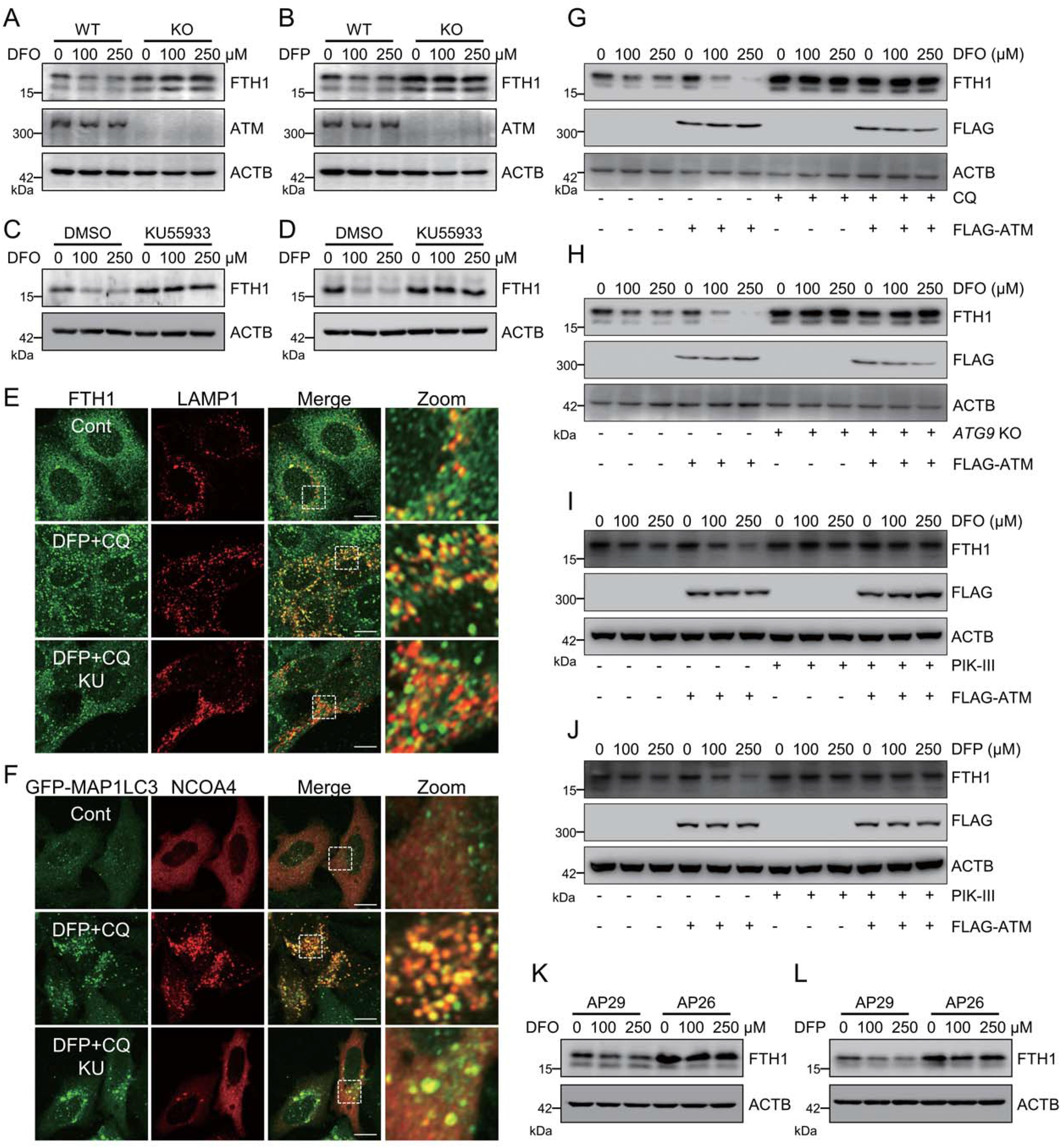
圖4:ATM主導(dǎo)鐵饑餓誘導(dǎo)的鐵自噬
5.ATM磷酸化鐵自噬受體NCOA4
穩(wěn)定的NCOA4通過FTH1的保守精氨酸殘基(Arg23)和NCOA4的C-末端結(jié)構(gòu)域之間的直接相互作用與鐵蛋白結(jié)合。在erastin補(bǔ)充期間,觀察到NCOA 4-FTH1相互作用的明顯增加,如通過免疫共沉淀分析所證明的(圖5A)。在平均孵育時(shí)間,通過使用泛磷酸化絲氨酸(p-Ser)抗體檢測(cè)到明顯的NCOA4磷酸化。重要的是,這種NCOA4磷酸化在erastin處理期間升高(圖5A)。因此,提出假設(shè)ATM激酶可能是負(fù)責(zé)這種NCOA4磷酸化。Erastin處理導(dǎo)致NCOA4磷酸化的升高,而當(dāng)與相應(yīng)的對(duì)照細(xì)胞相比時(shí),這種磷酸化在ATM敲除MEF細(xì)胞、ATM TRP53雙敲除AP26細(xì)胞和ATM敲低HT-1080細(xì)胞中幾乎完全被阻斷(圖5B-5D),表明ATM激酶對(duì)于響應(yīng)于Erastin的這種NCOA 4磷酸化是至關(guān)重要的。免疫共沉淀分析顯示S550中的突變幾乎消除了NCOA4磷酸化(圖5E)。為了進(jìn)一步證實(shí)進(jìn)化上保守的S550殘基(圖5F)是被ATM激酶磷酸化的關(guān)鍵位點(diǎn),通過用合成的NCOA4磷酸肽免疫小鼠,然后親和純化,產(chǎn)生針對(duì)磷酸化的S550的多克隆抗體。斑點(diǎn)印跡測(cè)定顯示,該抗體對(duì)磷酸肽(P-肽)的特異性比對(duì)照非磷酸肽(C-肽)高得多(圖5G)。通過使用該抗體,發(fā)現(xiàn)在對(duì)照HT-1080細(xì)胞中,響應(yīng)于erastin處理,S550處的NCOA4磷酸化升高。在穩(wěn)態(tài)和erastin處理狀態(tài)下,在ATM沉默的HT-1080細(xì)胞中觀察到S550處NCOA4磷酸化的顯著降低(圖5H)。總之,這些數(shù)據(jù)表明ATM在S550殘基處磷酸化NCOA4。
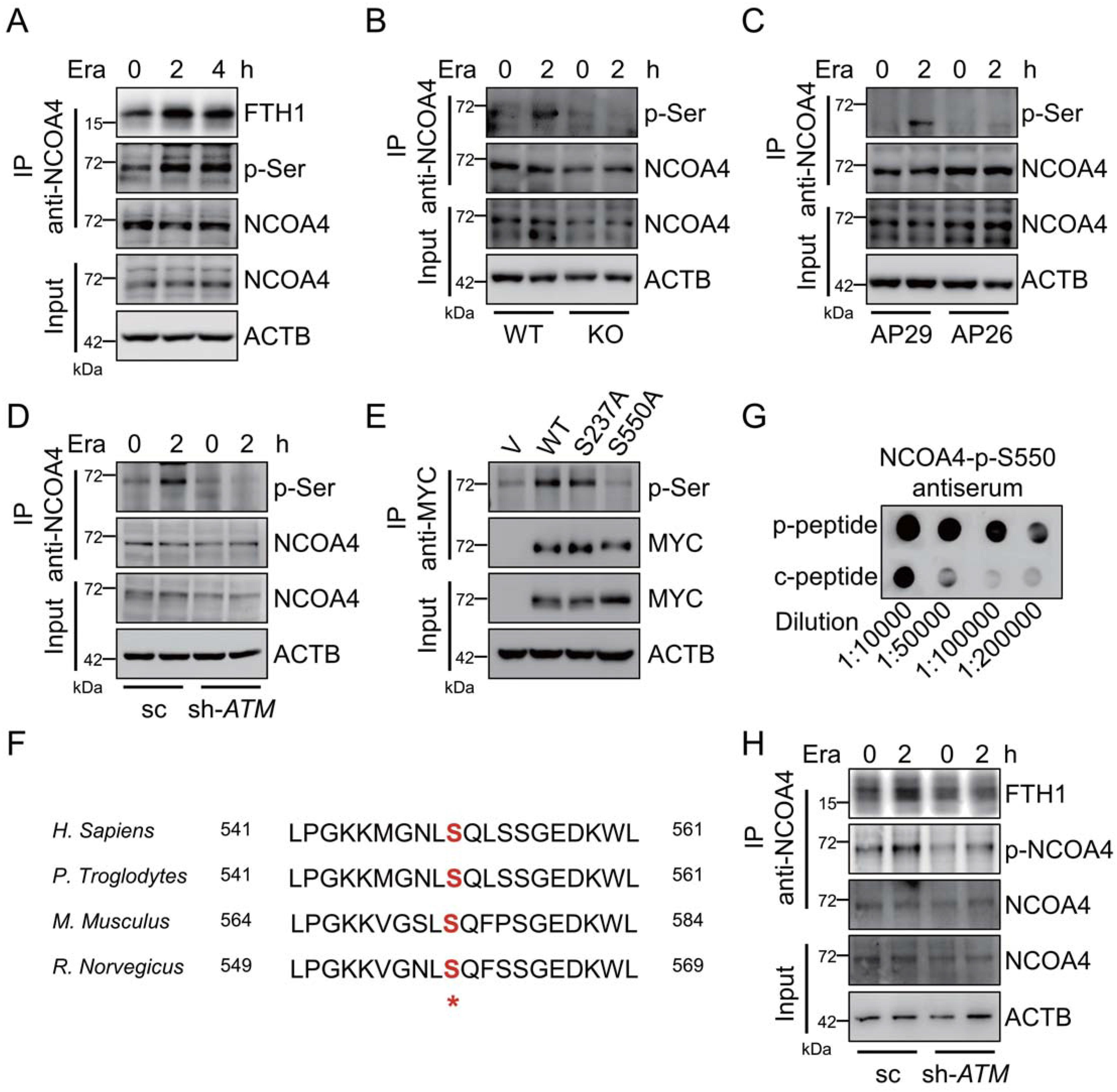
圖5:ATM磷酸化鐵自噬受體NCOA4
6.NCOA4磷酸化對(duì)鐵自噬和鐵死亡至關(guān)重要
ATM激酶介導(dǎo)的NCOA 4在S550殘基的磷酸化是否影響鐵自噬是下一個(gè)重要的問題。免疫共沉淀分析顯示,S550殘基中的突變明顯破壞了NCOA 4-FTH1相互作用(圖6A),表明ATM激酶在S550處的NCOAO4磷酸化可以增強(qiáng)NCOAO4與鐵蛋白的相互作用。為了進(jìn)一步闡明NCOAO4磷酸化在鐵自噬中的相關(guān)性,構(gòu)建了NCOA4穩(wěn)定敲低HT-1080(圖6 B)。與先前的研究一致,NCOA4沉默由于鐵自噬受損而升高FTHl水平(圖6 B)。此外,NCOA 4敲低深刻地消除了erastin誘導(dǎo)的FTHl增加,而該FTHl增加通過WT NCOA4的異位表達(dá)和S237A突變體而不是S550A突變體恢復(fù)(圖6 B)。鐵自噬可以更新鐵蛋白,補(bǔ)充細(xì)胞內(nèi)不穩(wěn)定鐵池。采用FerroOrange染色法檢測(cè)細(xì)胞內(nèi)不穩(wěn)定鐵的含量。NCOA4基因敲低后,Erastin誘導(dǎo)的熒光增強(qiáng)效應(yīng)減弱。一致地,WT NCOA4的異位表達(dá)和S237A突變體而不是S550A突變體,可以恢復(fù)NCOA4敲低的HT-1080細(xì)胞中的FerroOrange熒光增強(qiáng)(圖6C),表明在S550殘基處的NCOA4磷酸化對(duì)于與鐵死亡相關(guān)的鐵自噬和細(xì)胞內(nèi)生物活性鐵的升高是至關(guān)重要的。此外,在NCOA4敲低的細(xì)胞中,erastin誘導(dǎo)的鐵死亡被急劇消除,而WT NCOA4的異位表達(dá)和S237A突變體,而不是S550A突變體,可以恢復(fù)NCOA4敲低的HT-1080細(xì)胞中的細(xì)胞鐵死亡(圖6D和6 E)。類似地,WT NCOA4的異位表達(dá)而不是S550A突變體,可以恢復(fù)erastin誘導(dǎo)的脂質(zhì)過氧化反應(yīng),這顯著消除NCOA4敲低(圖6 F)。總之,這些數(shù)據(jù)表明,ATM激酶在S550殘基磷酸化NCOA4對(duì)于NCOA4鐵蛋白相互作用、鐵自噬介導(dǎo)的細(xì)胞游離鐵升高和隨后的鐵死亡執(zhí)行是至關(guān)重要的。

圖6:NCOA4磷酸化對(duì)于鐵自噬和鐵死亡至關(guān)重要
結(jié)論
綜上所述,這項(xiàng)研究表明了ATM激酶,DNA損傷反應(yīng)的主傳感器,是鐵死亡的關(guān)鍵上游調(diào)節(jié)因子。在機(jī)制上,ATM激酶磷酸化鐵自噬受體NCOA4,促進(jìn)NCOA4-FTH1相互作用以驅(qū)動(dòng)鐵自噬介導(dǎo)的鐵動(dòng)員并加劇脂質(zhì)過氧化。深入了解ATM激酶介導(dǎo)的鐵蛋白代謝的病理功能,有助于開發(fā)新的治療策略。
實(shí)驗(yàn)方法
細(xì)胞轉(zhuǎn)染和穩(wěn)轉(zhuǎn)株的構(gòu)建,CCK8實(shí)驗(yàn),流式細(xì)胞術(shù)檢測(cè)凋亡,脂質(zhì)過氧化物測(cè)量,GSH測(cè)定,qRT-PCR,western blot,免疫熒光實(shí)驗(yàn),鐵含量測(cè)定,核質(zhì)分離實(shí)驗(yàn),免疫沉淀實(shí)驗(yàn),克隆實(shí)驗(yàn)
參考文獻(xiàn)
Wu, H., Liu, Q., Shan, X., Gao, W., & Chen, Q. (2023). ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy, 19(7), 2062–2077. https://doi.org/10.1080/15548627.2023.2170960