






肝細胞癌的一區文章——分泌的蛋白酶PRSS35通過使CXCL2介導的中性粒細胞胞外陷阱來抑制肝細胞癌
肝細胞主要通過分泌調節細胞增殖、代謝和細胞間通訊的蛋白質發揮作用。在肝細胞癌(hepatocellular carcinoma,HCC)的進展過程中,肝細胞分泌蛋白質組作為腫瘤發生的結果和致病因素發生動態變化,盡管分泌蛋白質在這一過程中的全部功能尚不清楚。在這里,作者證明了分泌的偽絲氨酸蛋白酶PRSS35在HCC中作為腫瘤抑制因子發揮作用。在機制上,作者證明了有活性的PRSS35是通過前蛋白轉化酶切割加工的。活化的PRSS35通過靶向切割串聯賴氨酸(KK)識別基序抑制CXCL2的蛋白水平。因此,CXCL2降解減弱了中性粒細胞向腫瘤的募集和中性粒細胞胞外陷阱的形成,最終抑制了HCC的進展。這些發現拓展了作者對肝細胞分泌組在腫瘤發生發展中作用的理解,同時為PRRS35作為治療靶點或診斷生物標志物的臨床轉化提供了依據。本研究與2023年3月發表于期刊《Nature Communications》上,IF:16.6。
技術路線

主要研究結果
1、PRSS35蛋白豐度在HCC分泌組中降低
為確定與HCC發生和進展相關的潛在因素,作者對HCC分泌組進行了無標記蛋白質組學分析,并比較了條件培養基中分泌的人PLC肝癌細胞和人THLE3肝細胞的蛋白質譜。作者發現,與THLE3細胞相比,有236種分泌蛋白在PLC細胞培養的條件培養基中顯示出不同的豐度。PRSS35是PLC分泌組中下調最顯著的蛋白(圖1a)。使用識別蛋白N端的抗體進行Western blot(WB)分析PRSS35表明,相對于其在THLE3細胞中的積累,PLC、HepG2和Hep3B肝癌細胞內和細胞外PRSS35蛋白水平均顯著降低(圖1b)。有趣的是,富集在培養基中的PRSS35(SF-PRSS35)的分子量遠低于從細胞裂解液中分離的全長PRSS35(FL-PRSS35)(圖1b)。進一步利用針對PRSS35(即根據各自的抗原序列分別命名為N-PRSS35、M-PRSS35和C-PRSS35)不同肽段區域的抗體進行WB分析發現,在過表達PRSS35的PLC細胞培養基中富集到多個PRSS35蛋白短片段(圖1c)。質譜分析(MS)也證實,SDS-PAGE檢測到的較短波段確實包含了幾個PRSS35肽(圖1d)。總的來說,這些結果確定了PRSS35是一種分泌蛋白,在HCC細胞中豐度明顯較低,可能是由于裂解成多個片段。
隨后,作者發現與正常受試者相比,HCC患者血清中不同截短型PRSS35的水平顯著降低(圖1e),而全長PRSS35的水平無顯著變化。為進一步定量患者血清中的PRSS35水平,作者使用針對N末端區域的兩種抗體開發了定制的PRSS35 ELISA試劑盒。ELISA分析149例HCC患者和73例正常人血清,發現HCC患者血清PRSS35水平顯著低于正常人(圖1f)。這些結果表明分泌型PRSS35蛋白可以作為HCC患者潛在的預后生物標志物。此外,作者通過JASPAR數據庫預測可能調控PRSS35的轉錄因子,發現HNF4A是最有潛力的候選轉錄因子。此外,作者觀察到在HepG2細胞中敲低HNF4A后,PRSS35蛋白和mRNA水平均下調,而過表達HNF4A后,PRSS35蛋白和mRNA水平均上調。作者的數據表明HNF4A在肝癌中調控PRSS35的轉錄。
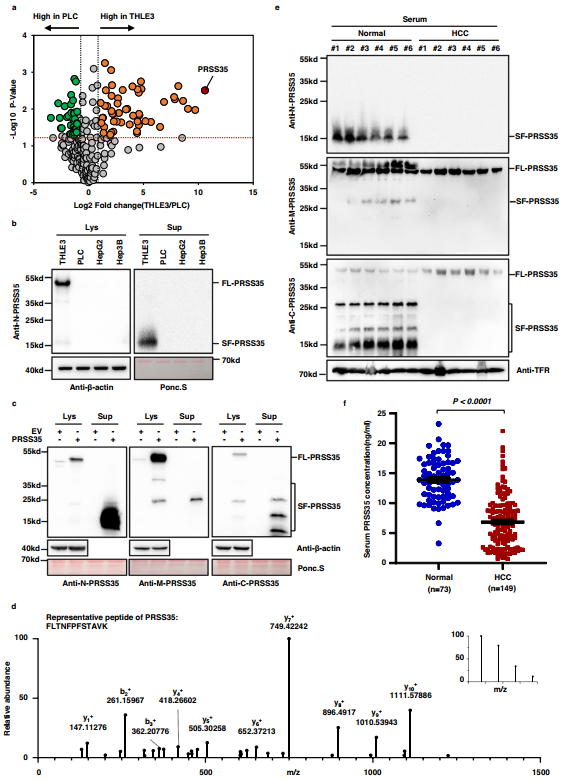
圖1 PRSS35是一種分泌蛋白,在HCC患者中減少
2、PRSS35是一種活性蛋白酶
PRSS35被注釋為無活性的假絲氨酸蛋白酶,因為在定義該酶家族的典型絲氨酸活性位點上存在蘇氨酸。然而,最近的研究表明,它的功能與卵母細胞成熟、受精和胚胎發育或腎小管間質炎癥有關,甚至調節傷口誘導的皮膚腫瘤發生。作者觀察到PRSS35降低與HCC之間的關聯,因此作者推測這種“假蛋白酶”可能表現出與活性絲氨酸蛋白酶相似的功能。為檢驗這種可能性,作者首先檢測了PRSS35蛋白酶對β-酪蛋白的活性,β-酪蛋白是許多絲氨酸蛋白酶的廣譜底物。為此,作者從大腸桿菌中純化帶有融合His標簽的全長PRSS35(FL-PRSS35),然后與β-酪蛋白孵育。作者觀察到在純化的FL-PRSS35存在下,β-酪蛋白被完全切割,表明PRSS35是一種活性蛋白酶(圖2a,左圖)。由于已知許多蛋白酶在發揮作用之前會被前蛋白轉化酶(proprotein converter,PC)切割而活化,因此作者在體外反應體系中對FL-PRSS35進行了WB分析。該分析揭示PRSS35存在多個截短形式,表明FL-PRSS35功能可能需要PC裂解成短的、有活性的形式(圖2a,右側面板)。
此外,作者通過對β-酪蛋白全長和裂解肽序列的LC-MS分析,確定了PRSS35在β -酪蛋白中的裂解位點。結果顯示,全長β -酪蛋白的肽段在K44前立即含有氨基酸殘基,但沒有切割β -酪蛋白(圖2b),說明K43和K44之間的位置是PRSS35的切割位點。
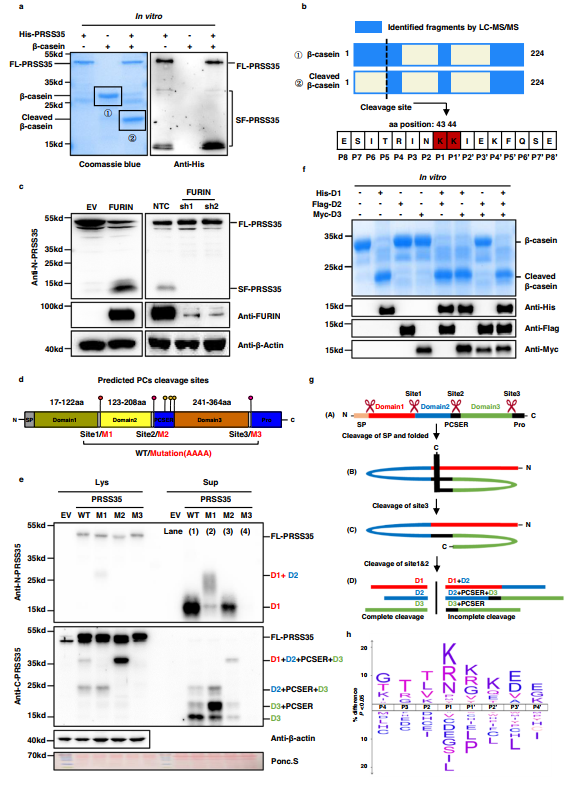
圖2 PRSS35被FURIN激活后發揮蛋白酶的功能
3、PRSS35被蛋白轉化酶激活
為研究PRSS35是否被PCs切割產生有活性的成熟體,作者研究了FURIN,一種廣為人知且普遍存在的PC對PRSS35切割的影響。作者構建了FURIN過表達和敲低的PLC細胞系,然后用WB進行檢測各系FL-PRSS35和SF-PRSS35水平。這些實驗表明,在FURIN過表達的情況下,FL-PRSS35水平降低,SF-PRSS35水平升高,而在FURIN敲低的細胞系中則觀察到相反的效果(圖2c),表明PRSS35被FURIN和潛在的其他PCs切割。對PRSS35蛋白序列進行生物信息學分析,發現人PRSS35至少存在6個潛在的PC切割位點。作者進一步預測來自其他物種的PRSS35的PC剪切位點,并在UniProt數據庫中找到了3個在所有物種中高度保守的剪切位點,包括小鼠和大鼠(圖2d)。值得注意的是,這3個額外的預測切割位點僅存在于人類PRSS35的site2附近區域,作者將其命名為PC切割位點富集區域(PCSER;圖2d)。
為確認PRSS35中預測的這三個高度保守的切割位點是真正的PC識別位點,作者分別在每個位點引入了非同義突變(AAAA)(圖2d)。這些突變破壞了PRSS35的切割,每個PRSS35突變體在WB分析中表現出不同的特異性條帶模式(圖2e)。首先,位點3(M3)的突變導致了所有SF-PRSS35的消失,表明M3對于所有PRSS35的切割是必需的,這意味著該位點的切割是后續蛋白水解加工(圖2e,泳道4與泳道1對比)所必需的。位點2(M2)突變導致由domain1(D1 , 12.4kd)或domain3(D3 , 14.2kd)組成的短型積累減少。此外,由結構域1、2、3和PCSER(即D1 + D2 + PCSER + D3)組成的中間片段增加了(圖2e ,泳道3與泳道1對比),這表明在位點2的剪切促進了隨后的PRSS35短形式的剪切。相反,位點1突變(M1)只抑制了導致domain1(D1 , 12.4kd)產生的剪切,并導致攜帶domain1和2(D1 + D2)(圖2e ,泳道2與泳道1的比較)的中間片段的積累。這些發現表明作者預測的切割位點是PRSS35上真正的PC識別位點,這些位點的切割對于PRSS35的成熟至關重要。
為確定哪種形式的PRSS35具有活性蛋白酶功能,作者從大腸桿菌中純化了His-D1、Flag-D2和Myc-D3融合蛋白片段。將這些純化的蛋白片段分別與β -酪蛋白孵育,發現β-酪蛋白在體外中只有D1存在時才被切割(圖2f),表明只有D1短形式具有活性。考慮到體外反應中呈現的短形式的分子量,作者的數據鞏固切割β-酪蛋白的是(12.4 kD)的D1短體,而不是FL-PRSS35(圖2a)。總之,這些結果表明PRSS35在被PC切割后發揮蛋白酶的作用,產生只含有D1的活性截短體。
為更好地了解這些預測的切割位點在PRSS35成熟過程中的作用模式和不同作用,作者進行了生物信息學分析,發現PRSS35的N端16個氨基酸很可能是信號肽。根據作者的WB數據,作者提出在N端的一個初始切割位點去除了FL-PRSS35(圖2g,構象A)的信號肽(SP),導致PRSS35的折疊構象,其中位點3暴露在蛋白表面,位點1和2埋在蛋白內部(圖2g,構象B)。PCs對位點3的切割改變了PRSS35的構象,暴露了位點1和位點2。然后,PCs可以被招募到鄰近的PCSER中的位點2和位點1(圖2g,構象C),最終導致PRSS35完全裂解為含有活性D1的短形式,以及含有D2、D3(圖2g,構象D)的非活性片段。
為進一步鑒定PRSS35識別的切割基序,確定其靶底物,作者采用了高通量蛋白酶篩(HTPS)。研究分析切割窗口發現,兩個相鄰的賴氨酸氨基酸(KK)作為PRSS35識別的核心切割基序(圖2h),這與作者在β -酪蛋白中鑒定到的切割位點一致(圖2b)。此外,作者合成了3條含有預測切割基序的熒光肽段(圖2h),發現His-D1肽段在體外中可以切割3條熒光肽段。這些發現證明PRSS35是一種由PCs裂解激活的胰蛋白酶樣絲氨酸蛋白酶,其活化的短形式D1可以通過靶向KK識別位點降解底物。
4、PRSS35在體內抑制HCC的發展
作者的數據表明,PRSS35是一種在肝癌中被抑制的活性蛋白酶,這促使作者研究PRSS35與腫瘤發生的關系。作者發現,與EV對照組相比,過表達小鼠PRSS35(mPRSS35)顯著抑制了Hepa1-6細胞在C57BL/6j小鼠中的生長(圖3a)。然后作者通過流體動力學注射質粒YAP-5SA41建立自發性HCC小鼠模型。結果顯示,mPRSS35過表達顯著抑制小鼠肝癌的發生(圖3b)。在YAP-5SA誘導的HCC小鼠模型中,作者觀察到與WT小鼠相比,mPRSS35-KO小鼠發生肝癌的速度加快(圖3c)。除了Hepa1-6小鼠HCC模型和YAP-5SA誘導的小鼠HCC模型外,作者還通過異種移植HCC模型證實了PRSS35在體內成瘤中的作用。作者發現人PRSS35(hPRSS35)過表達顯著抑制Balb/C純系裸鼠中HepG2腫瘤的生長(圖3d)。這些結果表明mPRSS35有助于小鼠的腫瘤抑制,盡管其在體外缺乏對HCC細胞生長的影響。
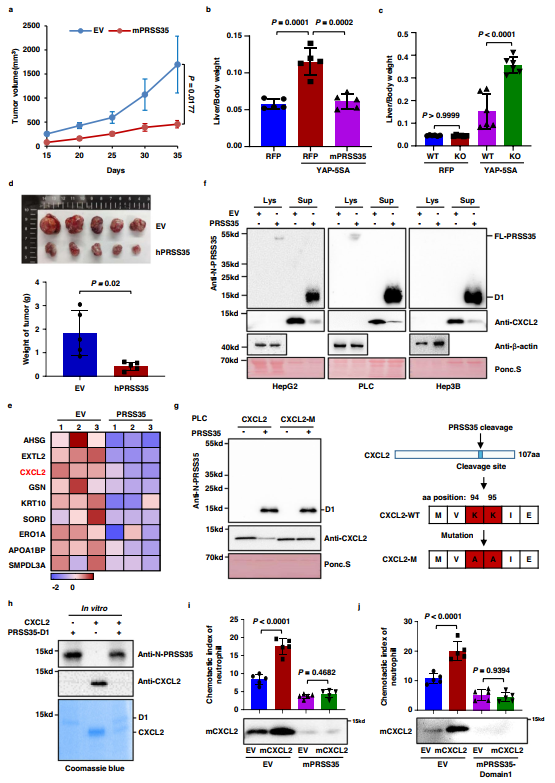
圖3 PRSS35通過降解CXCL2抑制中性粒細胞遷移
5、CXCL2是PRSS35的底物
為探索微環境中是否有某些因素影響PRSS35,作者結合RNA測序進行了SILAC蛋白質組學分析,以鑒定在PRSS35過表達PLC細胞的培養基中積累減少但轉錄水平不降低的分泌蛋白。該分析確定了9種分泌蛋白作為PRSS35的潛在底物(圖3e)。由于PRSS35的腫瘤抑制活性似乎依賴于體內微環境,鑒于CXCL2在腫瘤免疫微環境中作為趨化因子的作用,作者將研究重點放在了作為候選底物的CXCL2上。
WB分析表明過表達PRSS35顯著降低了HepG2、PLC和Hep3B細胞中CXCL2的胞外水平(圖3f)。此外,肽段序列分析表明CXCL2含有KK切割位點(圖3g,右),該基序的突變阻斷了PRSS35對CXCL2的切割(圖3g,左),進一步支持CXCL2是PRSS35的底物。為排除PRSS35降低CXCL2蛋白水平的其他間接機制,將E. coli純化的重組D1與E. coli純化的CXCL2蛋白共孵育,導致CXCL2降解(圖3h),證實CXCL2是PRSS35體外蛋白水解活性的直接底物。
由于CXCL2是趨化中性粒細胞的趨化因子,因此作者試圖確定PRSS35降解CXCL2是否可以抑制中性粒細胞的募集。對中性粒細胞遷移的分析顯示,從表達mCXCL2的細胞中收集的條件培養基增強了中性粒細胞的遷移,而過表達mPRSS35可以消除這種現象(圖3i,上)。與這些研究結果一致,WB顯示mPRSS35存在時,條件培養基中的mCXCL2蛋白顯著降解(圖3i,下)。對于PRSS35的活性D1形式也觀察到類似的結果(圖3j),從而證明mPRSS35介導的mCXCL2降解是中性粒細胞活性的促進因素。
5、PRSS35通過減少中性粒細胞向腫瘤的募集和減少NETs的形成來抑制HCC的進展
為研究PRSS35降解CXCL2在體內腫瘤進展中對中性粒細胞活性的影響,作者構建了穩定表達mPRSS35、mCXCL2和EV對照的Hepa1-6細胞系,并將這些細胞系分別接種到C57BL/6j小鼠皮下。結果表明,mCXCL2過表達促進了小鼠腫瘤的生長,而表達mPRSS35的小鼠表現出最低的腫瘤發生,有或無mCXCL2共表達(圖4a)。腫瘤組織的WB分析證實,在mPRSS35存在的情況下,mCXCL2蛋白被耗竭(圖4b)。進一步通過流式細胞術和IHC檢測腫瘤組織發現,在過表達mCXCL2的小鼠中,中性粒細胞向腫瘤內的募集增強,而與mPRSS35共表達后,中性粒細胞向腫瘤內的募集減弱(圖4c,e),表明mPRSS35介導的mCXCL2降解可以限制小鼠HCC組織中中性粒細胞的募集。
作者利用WB檢測腫瘤組織中NETs形成的標志物瓜氨酸化組蛋白H3(H3Cit)的水平,發現其在過表達mCXCL2的小鼠中顯著升高,但在過表達mPRSS35的小鼠中減弱(圖4b)。這些結果提示PRSS35和CXCL2在NETs形成過程中誘導了相反的作用。此外,作者通過ELISA檢測了荷瘤小鼠血清中H3Cit和另一個NETs形成的指標髓過氧化物酶- DNA(MPO-DNA)復合物。與mCXCL2促進H3Cit水平一致,作者發現mCXCL2過表達Hepa1-6細胞荷瘤小鼠血清中H3Cit和MPO-DNA復合物水平顯著升高,而mPRSS35過表達Hepa1-6細胞荷瘤小鼠血清中H3Cit和MPO-DNA復合物水平在mCXCL2過表達和mCXCL2過表達荷瘤小鼠血清中均無顯著變化(圖4d)。
在YAP-SSA誘導的HCC模型小鼠中,與上述在轉基因Hepa1-6誘導的HCC小鼠中得到的結果一致,作者發現與WT小鼠相比,mPRSS35-KO小鼠的腫瘤發展加快,而shRNA敲低mCXCL2(shmCXCL2)減弱了這種作用。此外,作者在荷瘤mPRSS35-KO小鼠的腫瘤組織和血清中觀察到更高的中性粒細胞浸潤和NETs形成,但在shmCXCL2的存在下被抑制(圖4g)。這些數據表明PRSS35可以通過降解CXCL2抑制腫瘤中性粒細胞浸潤和NETs形成。
為進一步研究中性粒細胞和NETs是否參與PRSS35介導的抑癌作用,將穩定表達空載體(EV)或Flag-mPRSS35的Hepa1-6細胞注射到C57BL/ 6J小鼠皮下。在細胞注射后第9天,作者將中性粒細胞中和Ly6G抗體或IgG對照以3天間隔注射到腫瘤和周圍皮下組織中。再次,與EV對照組相比,mPRSS35表達顯著抑制腫瘤生長,而在有或沒有Flag-mPRSS35過表達的腫瘤中,Ly6G處理導致類似的效果(圖4h)。與此一致,mPRSS35表達顯著抑制中性粒細胞腫瘤浸潤和NETs形成,Ly6G處理在Flag-mPRSS35過表達和不表達的腫瘤中產生類似的效果(圖4i-k)。結果表明,PRSS35通過抑制CXCL2介導的中性粒細胞募集和NETs形成來抑制HCC進展。
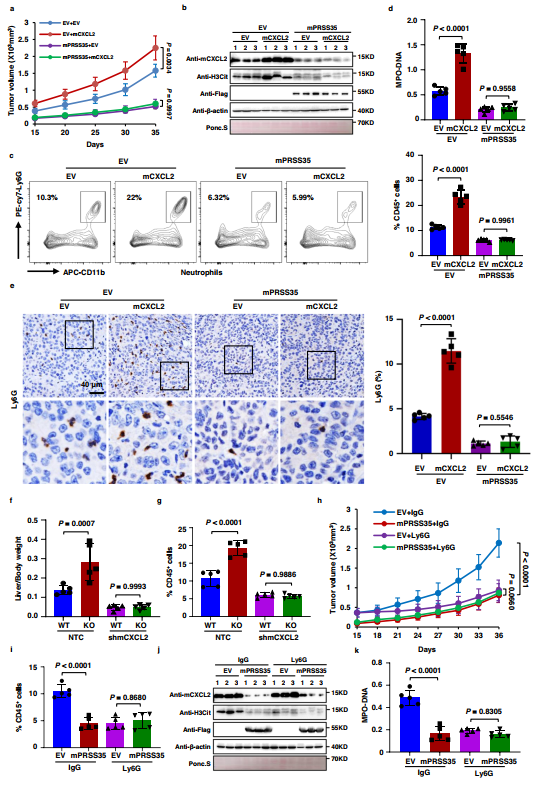
圖4 PRSS35通過抑制cxcl2介導的中性粒細胞NETs形成來抑制HCC的發展
實驗方法
人HepG2、Hep3B、PLC、Hepa1-6和HEK293T細胞培養,Western blot,ELISA法檢測血清PRSS35,凝膠內質譜,肝癌細胞系的生長曲線,高通量蛋白酶篩選,C57BL/6J小鼠,Balb/c nude老鼠,異種移植瘤模型,流式細胞術,臨床數據分析,血清MPO-DNA檢測,中心粒細胞隔離,中心粒細胞遷移檢測,PRSS35體外底物測定
參考文獻
Wang T, Zhou Y, Zhou Z, Zhang P, Yan R, Sun L, Ma W, Zhang T, Shen S, Liu H, Lu H, Ye L, Feng J, Chen Z, Zhong X, Wu G, Cai Y, Jia W, Gao P, Zhang H.(2023) Secreted protease PRSS35 suppresses hepatocellular carcinoma by disabling CXCL2-mediated neutrophil extracellular traps. Nat Commun.;14(1):1513. doi: 10.1038/s41467-023-37227-z.