






結直腸癌多標記轉錄分類反映腫瘤細胞群體的異質性
背景:轉錄分類已被用于將結直腸癌(CRC)分成具有不同生物學和臨床特征的分子亞型。然而,目前尚不清楚這些亞型是否代表著離散的、互斥的實體,還是具有潛在重疊的分子/表型狀態。因此,我們將重點放在CRC固有亞型(CRIS)分類器上,并評估將多個CRIS亞型分配給同一樣本是否提供額外的臨床和生物學相關信息。
方法:我們使用CRIS分類器的多標簽版本(multiCRIS)對新生成的606個CRC患者來源的異種移植物(PDXS)的RNA測序數據進行了分析,同時結合了人類CRC批量和單細胞RNA測序數據集。比較了單標簽和多標簽CRIS的生物學和臨床相關性。最后,開發了基于機器學習的多標簽CRIS預測器(ML2CRIS)用于單個樣本的分類。
結果:令人驚訝的是,約一半的CRC病例可以明顯地分配給多個CRIS亞型。單細胞RNA測序分析揭示,多個CRIS成員身份可能是由于同時存在不同CRIS類別的細胞,或者較少情況下由于具有混合表型的細胞。發現多標簽分配可以改善對CRC預后和治療反應的預測。最后,ML2CRIS分類器在單個樣本分類的情境下被驗證具有相同的生物學和臨床相關性。
結論:這些結果表明,即使同時分配給同一CRC樣本,CRIS亞型仍保留其生物學和臨床特征。這種方法有潛力在其他癌癥類型和分類系統中推廣應用。
該研究于2023年5月發表發表在《Genome medicine》,IF:15.266。
技術路線:
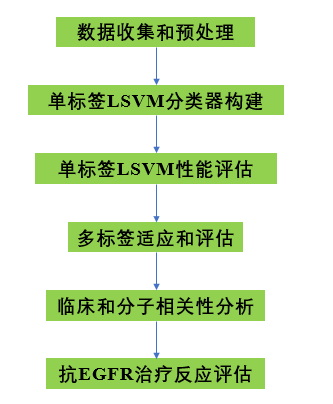
實驗方法:異種移植物收集、TCGA和PDX RNA-SEQ數據預處理、CRC單細胞數據及其預處理、bulk/scRNA-seq數據和預處理、scRNA-seq數據的偽批量、CRIS分類、單標簽分級機、多標號單樣分級機。
1、結直腸癌內在亞型的多標簽CRIS分層研究
為了改善結直腸癌的分層,并根據CRIS分類捕捉生物特征,我們推斷其最近模板預測(NTP)算法不僅可以用于指定最顯著的單一類別,還可以評估每個樣本對所有CRIS類別的分配,以及每個分配的虛假發現率。因此,我們實施了基于NTP的CRIS分類器的新的多標簽版本,名為“multiCRIS”,能夠根據與每個CRIS中心點的距離和其顯著性將每個樣本分配給一個或多個CRIS類別。
首先,將MultiCRIS應用于來自癌癥基因組圖譜(TCGA)的620個樣本的RNA測序數據集,以明確地將91%的樣本至少分配給一個類別(圖1a)。有趣的是,52%的樣本還可以被確信地分配給其他CRIS亞型(圖1b)。
值得注意的是,對于所有的CRIS亞型,次要分配的數量與主要分配大致相等(圖1c)。多重分配主要發生在兩個特定的亞家族之間:CRISA/CRIS-B和CRIS-C/CRIS-D/CRIS-E。最后,為了評估這些多重分配是否捕捉到具有多個CRIS生物特征的腫瘤,我們探索了與每個CRIS類別相關的主要特征。
有趣的是,分配給次要類別的樣本在圖1d中顯示了類別的關鍵分子特征,包括CRIS-A中的MSI狀態,CRIS-C中的KRAS突變的消失,以及CRIS-D/CRIS-E中的WNT信號通路活性和CRIS-B樣本中的上皮間質轉化(EMT)。值得注意的是,我們觀察到具有多個分配的樣本傾向于與CRIS中心點之間的距離較大,這可能反映了同時具有不同表型的細胞組成或具有不同表型的細胞混合的情況。

圖1展示了針對TCGA數據集中的596個結直腸癌樣本進行的多標簽CRIS分類的結果。
2、多個CRIS分配中的單細胞異質性。
觀察到一部分結直腸癌的多個類別分配可以通過兩種方式解釋:腫瘤由具有模糊表型的癌細胞組成,或者存在混合的不同亞型細胞群體。為了探索支持多個CRIS分配的異質性,我們在一個由PDXS(患者源性異種移植)衍生的5個結直腸癌器官樣本集合中進行了一系列的配對單細胞RNA測序(scRNA-seq)和批量譜分析。這些數據允許直接比較單細胞和批量轉錄組譜分析結果。作為第三個選擇,通過聚合一個樣本中所有單細胞譜分析結果來獲得偽批量譜分析結果。值得注意的是,盡管來自單個細胞的譜分析結果平均捕獲了至少5個支持讀數的1116個轉錄本,但偽批量譜分析結果平均涵蓋了超過17,095個轉錄本。如預期的那樣,匹配的批量/偽批量樣本的譜分析結果顯示了強烈的相關性,而無法通過非匹配比較獲得。這些結果表明,(i)單細胞譜分析結果顯示出高度的異質性,以及(ii)聚合的單細胞譜分析結果能夠重現批量譜分析結果中所獲得的轉錄組譜。因此,這種3D體外器官樣本培養系統捕獲了具有復雜轉錄組異質性的細胞譜。
值得注意的是,我們發現存在同時存在的細胞混合物,每個混合物具有一個單一的CRIS分配,以及具有混合多個CRIS亞型的細胞。來自給定器官樣本的個別細胞主要被分配到該器官樣本的批量譜分析結果所定義的CRIS亞型/亞型組(圖2)。
這些結果強調了在單細胞分辨率下,大多數細胞被分配到單個CRIS亞型,并且它們的混合導致了批量轉錄組的多亞型分配;然而,也有可能存在一小部分具有混合表型的細胞,在給定的批量樣本中對多個CRIS亞型的分配產生貢獻。事實上,在所有接受多個CRIS批量分配的器官樣本中,我們檢測到了具有不同CRIS標識的細胞和具有混合表型的細胞的共存(圖2)。
為了將我們的觀察擴展到人類腫瘤,我們利用來自一組患者的公共單細胞RNA測序數據(GSE132465),重點關注上皮細胞,比較偽批量和單細胞的多標簽CRIS分配情況:這種分析證實了存在多個CRIS分配的患者。在這些樣本中,我們證實大多數單個細胞被分配到特定的CRIS亞型(64%的分類細胞,其中75%被分配到單個CRIS亞型,25%被分配到多個CRIS組;圖3a)。然而,類似于器官樣本,每個樣本由不同的細胞群體組成,這些細胞群體被分到不同的CRIS亞型中,導致了一個復雜的表型,該表型通過偽批量分析的多個CRIS分配被捕捉到(圖3b)。因此,被分配到單個CRIS亞型的樣本往往具有更高比例的被分配到該亞型的細胞。在特定樣本中,具有多標簽分配的單個細胞的高百分比可能反映出組織中正在經歷功能轉變或穩定的中間分化階段。例如,在患者SMC17中發生了這種情況(圖3b),其中57%的分類細胞顯示出多標簽表型。類似地,SMCO3和SMC21患者分別顯示出34%和28%的具有混合表型的細胞(圖3b),與它們在批量分析中追蹤到的多標簽狀態一致。
總的來說,這些結果表明,CRIS轉錄組的異質性根源于單個細胞水平,而單個細胞的表型總結起來定義了腫瘤批量的CRIS分類。因此,多CRIS腫瘤的證據主要可以通過具有特定功能特征的不同細胞群體的鑲嵌組成或具有混合表型的少量混合細胞來解釋。
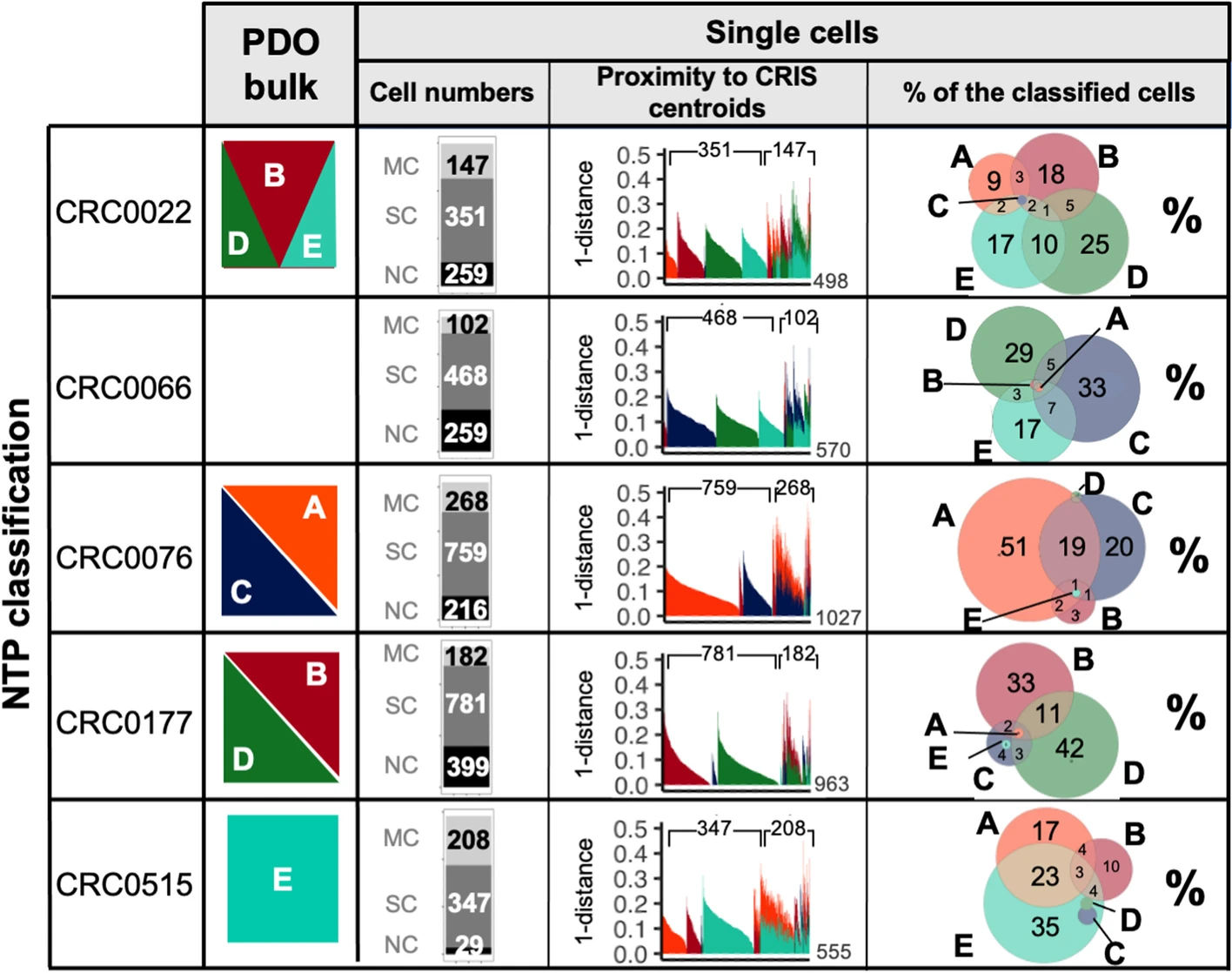
圖2展示了人類器官樣品的MultiCRIS分類結果。

圖3展示了人類結腸直腸癌(CRC)的MultiCRIS分類結果。
3、針對CRIS分類的單樣本方法
MultiCRIS為復雜的生物學和臨床結果鋪平了道路。然而,它受到其NTP實現的影響,該實現依賴于在樣本批次上計算的質心距離和基因級別的Z分數,不允許單個樣本的分類。為了克服這個問題,我們轉向單樣本算法,能夠獨立地對每個樣本進行分類:這些算法既可以處理只對主要類別進行單標簽分配的情況(SC),也可以處理多標簽分配的情況(MC),以捕捉內部的異質性。我們的工作流程如圖4所示;它包括對所有算法進行初始訓練階段(藍色),在測試數據上對它們的性能進行評估(粉色),以及對最有希望的單樣本方法進行最終的臨床和生物驗證(綠色)。我們首先實現了單樣本單標簽算法,能夠識別每個樣本的最顯著(主要)類別。考慮的方法包括隨機森林(RF)、線性支持向量機(LSVM)、多項式(PSVM)和高斯徑向基函數(GRBF-SVM)核的支持向量機、神經網絡(NN)和極端梯度提升樹(XGBoost)。隨后,通過提取每個樣本的所有CRIS類成員關系,TESE單樣本算法被適應于多標簽上下文;這使得能夠對照多標簽NTP(多CRIS)的結果來驗證其結果。
為了評估每個算法并確定最適合在臨床應用的單樣本分類器中預測CRIS類別成員資格的算法,我們利用了來自TCGA項目的原發性結直腸癌樣本集(n=562)和來自患者衍生的異種移植瘤(PDXS,n=550)的隊列。TCGA數據被分成訓練集和測試集,保持整個數據集中CRIS類別比例不變。每個分類器僅考慮CRIS基因的表達值作為特征空間,并使用NTP主類作為目標參考進行訓練,無論其是單標簽還是多標簽的使用。TCGA樣本的30%和完整的PDXS數據集被用作兩個獨立的測試集,以評估單標簽和多標簽分類器的結果。
在針對主要類別分配的單標簽評估中,我們使用全局準確率、精確度和召回率,衍生的度量指標(F1分數和馬修斯相關系數(MCC)),以及基于閾值的度量指標(接收器操作特性曲線下面積和精確率-召回率曲線下面積),以評估所考慮算法的性能,并將其與原始的NTP方法的性能進行比較。LSVM在TCGA測試集上達到了約80%的準確率,在PDXS上達到了75%的準確率,在考慮了所有類別的精確度和召回率時表現良好(圖5)。雖然XGBoost和RF取得了有趣的表現,但它們的類別特定行為似乎不太穩定,并且整體上略遜于LSVM(圖5)。
此外,在單標簽情況下,機器學習方法與TSP方法進行了比較,TSP方法是單樣本CRIS分類器的首次嘗試,最初顯示出與NTP分類的相當有限的一致性。本研究中證實了其次優結果:我們所有的分類器都比TSP獲得了更高的準確性,最高準確率為70.7%。特別是對于LSVM來說,即使在TCGA測試集上F1分數最低的類別(CRIS-E,為73%)也顯著超過了TSP的結果(CRIS-E為28%)。
在PDXS數據集上,LSVM取得了最令人信服的結果。在TCGA中,CRIS-A是最常見的類別,略多于CRIS-C;其次是CRIS-E,而CRIS-B和CRIS-D的規模較小但可比較。在PDXS中幾乎可以觀察到相同的趨勢,例外就是CRIS-A類在PDXS中的表示較少,這是因為來自轉移性結直腸癌的樣本中MSI病例(CRIS-A富集)的稀缺性。
因此,基于性能評估,我們確定LSVM作為最佳的單標簽分類器,用于預測每個單個CRC樣本的主要CRIS類別。然而,所有三種訓練過的算法都能通過算法自適應技術計算出對所有5個CRIS類別的隸屬度,為多標簽情景奠定了基礎。

圖4展示了基于機器學習構建單樣本CRIS分類器的工作流程。

圖5展示了基于機器學習的CRIS分類器的性能評估結果
4、通過單樣本方法,進行多標簽CRIS分類。
在算法自適應策略的基礎上,我們開發了多標簽適應(mla)的單樣本CRIS分類器。具體來說,每個mla算法從其單標簽版本繼承了主要類別的分配能力,但可以將任何異質樣本與一個或多個額外的次要類別關聯起來。
為了評估mla分類器,我們使用了與單標簽類似但適應于多標簽環境的度量指標(放寬準確率、精確率、召回率),以及特定的多標簽度量指標(平均精確率、Hamming損失、子集準確率和多標簽準確率)。所有這些指標都將mla算法的結果與本研究中引入的MultiCRIS方法獲得的目標分配進行比較。
在mla算法中,LSVM在考慮類別精確率和召回率時仍然達到了最佳整體性能,顯示出在多標簽環境中也是最穩健的方法(圖5)。此外,LSVM在91.7%的情況下分配了主要的多標簽CRIS類別(即根據NTP算法確定的最突出的類別),并且在預測TCGA測試樣本的多標簽特征時達到了92.6%的平均精確度。在考慮Hamming損失時,即錯誤分類標簽的平均比例,LSVM在TCGA測試集和PDXS集中都具有最低的損失比例。最后,LSVM的子集準確率(嚴格相同的標簽歸屬)也非常重要,尤其考慮到每個算法只通過提供主要類別作為參考目標進行訓練。
因此,LSVM顯然是在單標簽或多標簽視角下執行單樣本分類的最佳方法。
5、單標簽和多標簽LSVM分類器的臨床和生物學評估
我們首先評估了基于LSVM模型在TCGA數據集上的預后價值,考慮了不同的情景:僅考慮作為主要類別分配的樣本、僅考慮作為次要類別分配的樣本,或者考慮所有被分類為CRIS類別的腫瘤,不論其是主要還是次要分配。在所有這些情況下,使用Fisher檢驗進行的任何比較都是針對那些完全未被分配到所研究的CRIS類別的樣本。對于基于NTP和LSVM的單標簽分類器,Kaplan-Meier(KM)生存分析證實了CRIS-B類與不良預后的顯著關聯(圖6a、b)。有趣的是,通過多標簽分配,將次要CRIS-B分配的樣本從非CRIS-B組中排除,突出了與不良預后的更高的相關性(圖6c、d)。因此,當排除具有主要CRIS-B分配的樣本進行分析時,次要CRIS-B分配的樣本顯示出更差的預后(圖6e、f)。當將主要和次要CRIS-B病例合并時,預后顯著性達到最大值(圖6g、h)。值得注意的是,在所有情況下,基于LSVM的分類器具有更高的預后顯著性。
在PDXS隊列中評估了對抗EGFR治療的反應。我們確認了CRIS-C與Cetuximab的敏感性相關聯,其中單標簽LSVM(優勢比(O.R.)= 3.281,置信區間(CI)= 1.66-6.73)和多標簽LSVM,包括在CRIS-C隊列中的次要分配,顯示出類似的表現(O.R. = 3.36,CI = 1.24-10.64)。
所有這些證據都證實了我們的LSVM單樣本模型的預測結果的可靠性,特別是mla LSVM,即ML2 CRIS(多標簽機器學習CRIS)。ML2 CRIS能夠突出樣本的生物學內在異質性(如果有的話),同時在臨床應用中對每個患者進行個體化評估。這證明了ML2 CRIS在臨床使用環境中的可靠性。
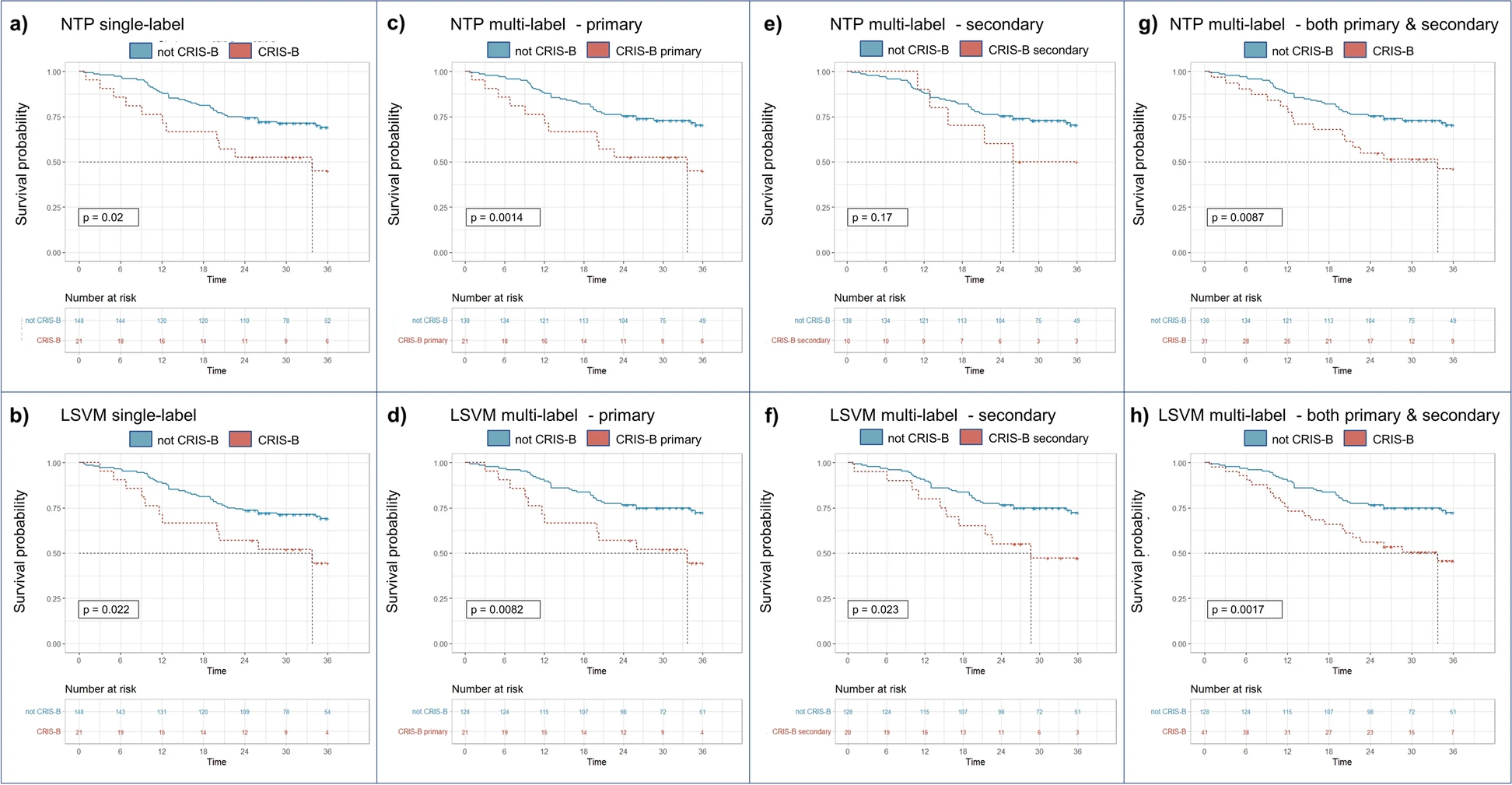
圖6展示了單標簽和多標簽CRIS-B分類在預后方面的意義
參考文獻:
Cascianelli, S., Barbera, C., Ulla, A.A. et al. Multi-label transcriptional classification of colorectal cancer reflects tumor cell population heterogeneity. Genome Med 15, 37 (2023).https://doi.org/10.1186/s13073-023-01176-5