






抑制HDAC3通過增強CXCL10介導的趨化性和免疫細胞募集促進抗腫瘤免疫
實驗方法:細胞增殖和活力分析,組織分離,免疫熒光染色,ELISA,ELISpot (酶聯免疫斑點測定法),腫瘤浸潤性CD4+和CD8+ T細胞的大量RNA測序及分析,腫瘤RNA測序及分析,qRT-PCR,ChIP-seq和ChIP-qPCR檢測,流式細胞術,Western blot,TMA的IHC測量與分析
組蛋白去乙酰化酶(histone deacetylase, HDAC)抑制劑是一類新的抗癌藥物,一般認為其抗腫瘤活性是通過直接引起腫瘤細胞周期阻滯和細胞凋亡來發揮的。然而,我們證明了I類HDAC抑制劑,如Entinostat和Panobinostat,可以有效抑制免疫正常而非免疫缺陷小鼠的腫瘤生長。對Hdac1, 2, 3敲除腫瘤細胞的進一步研究表明,HDAC3的腫瘤特異性失活通過激活抗腫瘤免疫來抑制腫瘤生長。具體來說,我們發現HDAC3可以直接結合到啟動子區域,抑制CXCL9, 10, 11化學因子的表達。Hdac3缺陷的腫瘤細胞高水平表達這些趨化因子,通過將CXCR3+ T細胞募集到腫瘤微環境(TME)中,抑制免疫能力小鼠的腫瘤生長。此外,HDAC3與CXCL10在肝細胞癌腫瘤組織中的表達呈負相關,也提示HDAC3可能參與抗腫瘤免疫調節和患者生存。因此,抑制HDAC3通過增強免疫細胞向TME的浸潤來抑制腫瘤生長。這一抗腫瘤機制可能有助于指導基于HDAC3抑制劑的治療。
技術路線:

結果:
(1) HDAC抑制劑以免疫依賴的方式抑制腫瘤生長
HDAC抑制劑已被廣泛用于癌癥治療。其中,Panobinostat(也稱為LBH589,一種泛HDAC抑制劑)和Entinostat(也稱為MS275, HDAC1和3選擇性抑制劑)正在進行III期臨床試驗。我們首先檢測了恩替諾他和帕比諾他對MC38小鼠結腸腺癌和MCA205纖維肉瘤腫瘤生長的影響。我們發現Entinostat和Panobinostat在免疫正常的C57BL/6小鼠中有效抑制MC38和MCA205腫瘤的生長(圖1A-D),但在免疫缺陷的裸小鼠中無效(圖1E-H)。這些結果表明,I類HDAC抑制劑Entinostat和Panobinostat以免疫依賴的方式抑制MCA205和MC38腫瘤的生長。
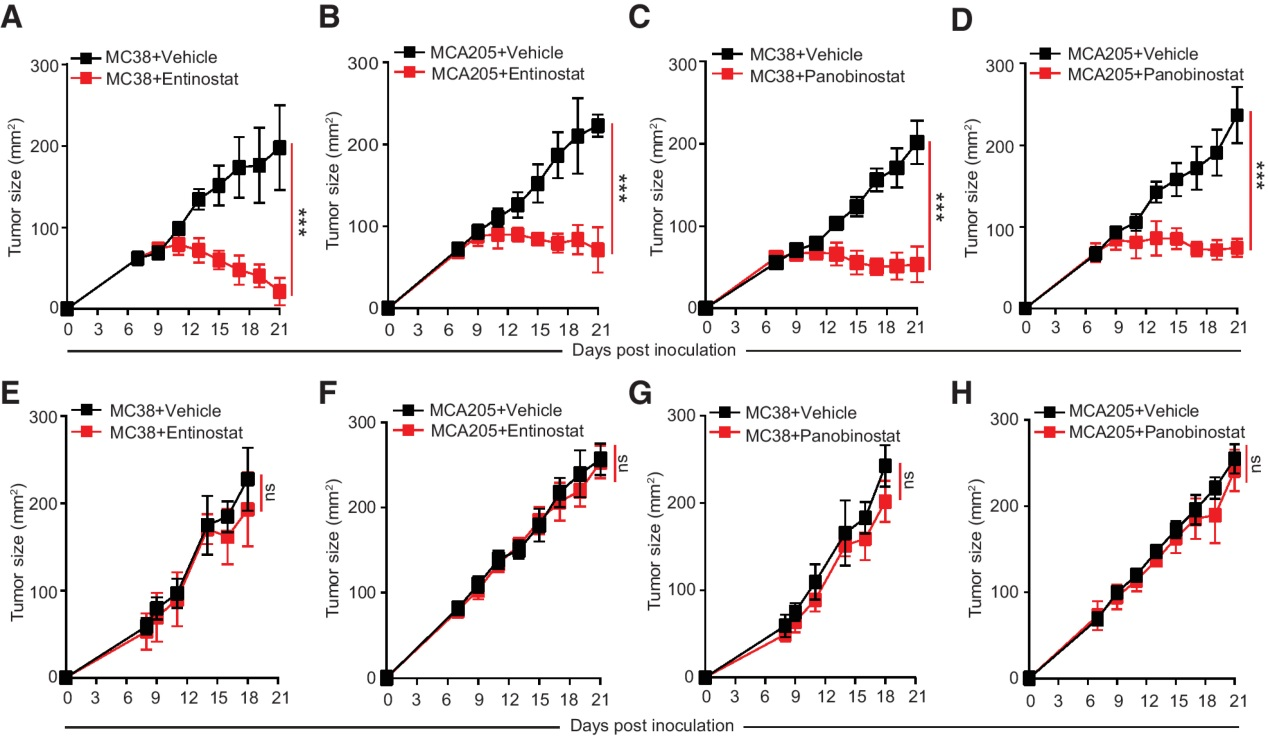
圖1:HDAC抑制劑以免疫依賴的方式抑制腫瘤生長
(2) Hdac3缺乏抑制腫瘤生長,而腫瘤生長依賴于宿主免疫反應
Panobinostat和Entinostat均抑制MCA205和MC38腫瘤的生長,提示抑制一種或多種I類HDAC可能抑制腫瘤生長。為了探索Hdac1, 2, 3基因在腫瘤生長中的作用,我們利用CRISPR/Cas9技術敲除MC38細胞中的Hdac1, 2, 3基因(補充圖S1A-S1C),并將細胞注射到C57BL/6小鼠體內。比較Hdac1?/?,Hdac2?/?和Hdac3?/?我們發現Hdac3缺失顯著抑制MC38腫瘤的生長,而Hdac1和Hdac2缺失對MC38腫瘤的生長無明顯影響(圖2A)。為了證實Hdac3基因在腫瘤生長中的作用,我們進一步敲除(Hdac3+/?)和敲除(Hdac3?/?) MC38和MCA205細胞中的Hdac3基因。比較Hdac3+/?和Hdac3?/?我們發現,在C57BL/6小鼠和裸鼠中,敲低和敲除MC38和MCA205腫瘤及其相應的WT腫瘤均顯著抑制了C57BL/6小鼠中MC38和MCA205腫瘤的生長(圖2B和C),而在裸鼠中則沒有抑制作用(圖2D和E)。這些結果表明,HDAC3促進腫瘤生長并依賴于宿主免疫應答。
將MCA205細胞靜脈注射到小鼠體內后,腫瘤會發展并轉移到多個器官,導致小鼠死亡。然后我們比較靜脈注射WT或Hdac3?/?的C57BL/6小鼠的存活率。MCA205細胞。移植WT MCA205細胞的小鼠在移植后不能存活超過17天,而所有移植Hdac3?/? MCA205細胞存活(圖2F),表明Hdac3缺失減少了轉移并保護小鼠免于轉移相關死亡。我們還分析了GEO和TCGA數據庫中876例胃癌患者Hdac1, 2, 3基因表達與OS的相關性。Hdac3基因的高表達與胃患者較低的OS相關,而Hdac1和Hdac2基因的高表達與胃患者較高的OS相關(圖2G-I)。此外,HDAC3 mRNA在各種癌癥組織中的表達均高于相應的正常組織,包括膽管癌、宮頸鱗狀細胞癌和宮頸內膜腺癌、肺腺癌、肺鱗狀細胞癌(補充圖S2)。這些結果提示特異性抑制Hdac3可能通過抗腫瘤免疫抑制腫瘤生長。

圖2:Hdac3缺乏抑制腫瘤生長并依賴于宿主免疫反應
(3) Hdac3缺陷腫瘤的局部免疫反應增強
CCK-8 (補充圖S3A和S3B)、Ki-67染色(補充圖S3C和S3D)和單克隆形成(補充圖S3E和S3F)檢測顯示,Hdac3缺乏對MC38和MCA205細胞的活力和增殖沒有影響。為了探索HDAC3是否在調節局部TME中發揮作用,我們使用流式細胞術檢測分析了WT和Hdac3+/? MCA205腫瘤中的腫瘤浸潤淋巴細胞(TIL),因為Hdac3?/?細胞沒有形成足夠大的腫瘤進行流式細胞術分析。事實上,我們觀察到腫瘤浸潤的CD4+、CD8+、CD11c+、CD11b+F4/80+、CD4+ IFNγ+和CD8+ IFNγ+免疫細胞的數量增加。MCA205腫瘤(圖3A;補充圖S4)。結果表明,當腫瘤細胞特異性缺乏Hdac3時,更多抗原呈遞DCs和腫瘤殺傷T細胞被招募到MCA205 TME中。為了更好地計數腫瘤浸潤T細胞的大小和分布,對WT、Hdac3+/?和Hdac3?/? MCA205腫瘤進行免疫熒光染色。更多的CD4+、CD8+和IFNγ+細胞浸潤Hdac3+/?和Hdac3?/? MCA205腫瘤與WT型MCA205腫瘤的比較(圖3B, C;補充圖S5A和S5B)。還采用ELISpot法評估WT和Hdac3+/? MCA205穩定表達的雞卵細胞中的OVA特異性IFNγ分泌T細胞的頻率。結果顯示,Hdac3+/? MCA205腫瘤中IFNγ分泌T細胞比WT腫瘤中更多(圖3D和E)。這些結果表明,Hdac3缺陷腫瘤中浸潤T細胞的數量和特征明顯不同于WT腫瘤。
為了表征TME中浸潤性T細胞的特征,我們通過FACS從WT和Hdac3+/? MCA205腫瘤中分離CD45+CD4+ and CD45+CD8+ T細胞,并進行大量RNA測序檢測基因表達。與WT細胞相比,從Hdac3+/?腫瘤分離的T細胞表達更高水平的轉錄因子,如Eomes和Tcf712;細胞表面受體如Sell、Vsir和Cd28;效應分子如Gzmb、Ifng、Prf1、Tnf;和耐受性相關基因如Nr4a2和Egr2 (圖3F)。以上結果表明,腫瘤細胞特異性Hdac3缺失引發了免疫細胞的差異浸潤,在TME中比在WT腫瘤中更強的局部免疫反應。
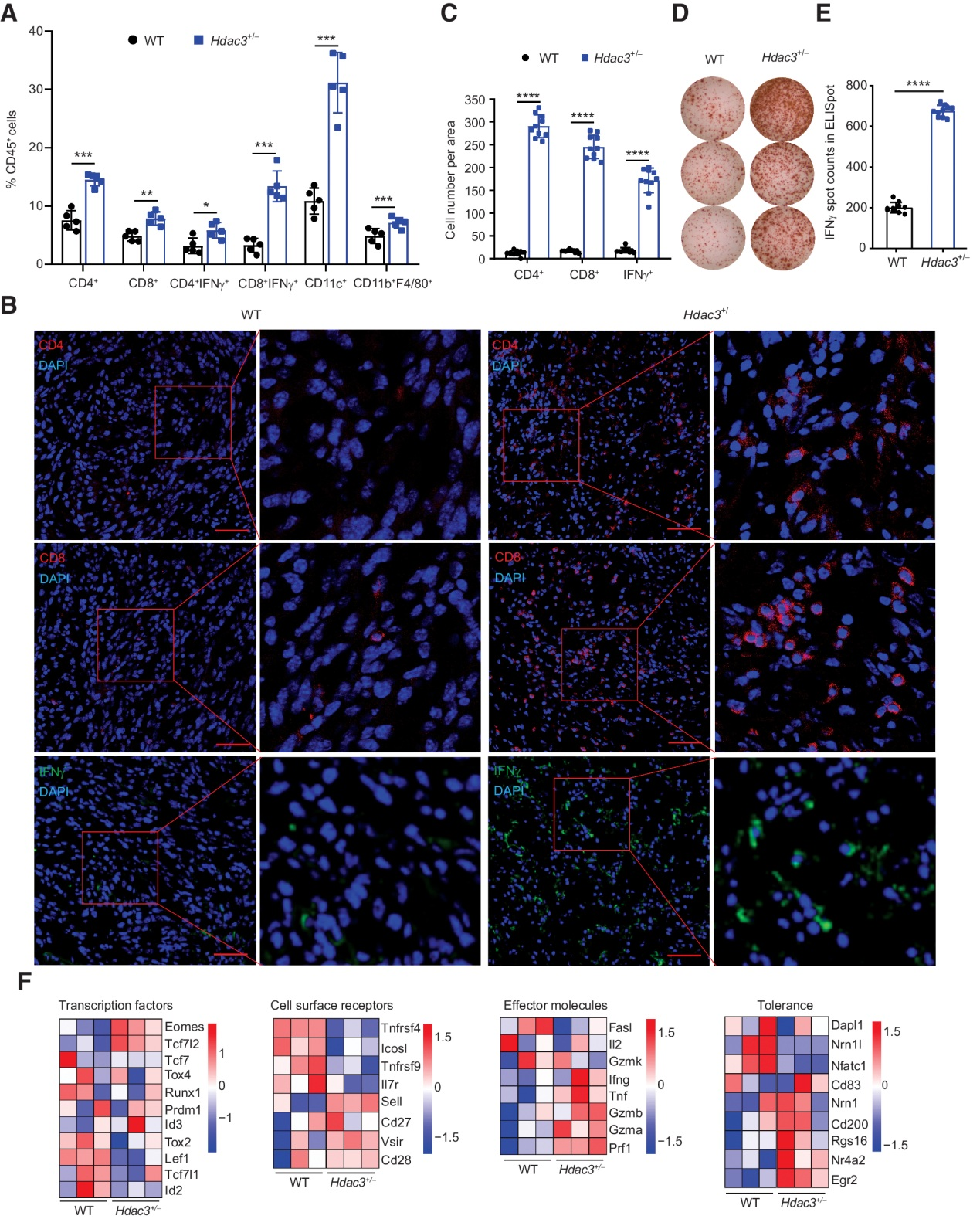
圖3:Hdac3缺陷腫瘤的局部免疫反應增強
(4) Hdac3缺乏上調趨化因子信號通路和T細胞受體信號通路
大量RNA-seq結果(圖3F)顯示,HDAC3可能通過調節TME中免疫細胞(尤其是T細胞)的浸潤和特征來影響腫瘤生長。為了探索HDAC3調節T細胞浸潤和TME的機制,我們對WT和Hdac3+/? MCA205腫瘤組織進行RNA測序。KEGG注釋分類分析比較Hdac3+/?和WT腫瘤顯示,Hdac3缺失顯著上調趨化因子信號通路、T細胞受體(TCR)信號通路和細胞因子-細胞因子受體相互作用過程中的基因(圖4A和B)。GSEA還顯示,趨化因子信號通路(圖4C)、細胞因子-細胞因子受體相互作用(圖4D)和TCR信號通路(圖4E)在Hdac3缺失的MCA205腫瘤中顯著富集。這些結果表明,與T細胞募集相關的TCR信號通路和趨化因子信號通路可能有助于抑制Hdac3缺陷MCA205細胞的腫瘤生長。然后,我們選擇了與T細胞和T細胞募集相關的基因,并評估了它們在WT和Hdac3+/? MCA205腫瘤中的相對表達。豐富的T細胞標記物(Cd3d、Cd3e、Cd3g、Cd4、Cd8a、Ctla4、Zap70和Cxcr3)和T細胞募集相關基因(Cxcl9, 10, 11)在Hdac3缺陷腫瘤中顯著上調(圖4F)。為了驗證RNA測序的結果,通過qRT-PCR將Hdac3?/? MCA205腫瘤與WT腫瘤中的基因表達進行比較。我們證實,Hdac3缺陷導致T細胞標記和T細胞招募相關基因上調(補充圖S5C)。這些結果使我們假設HDAC3可能通過降低趨化因子如Cxcl9, 10, 11的表達來抑制T細胞募集到TME。
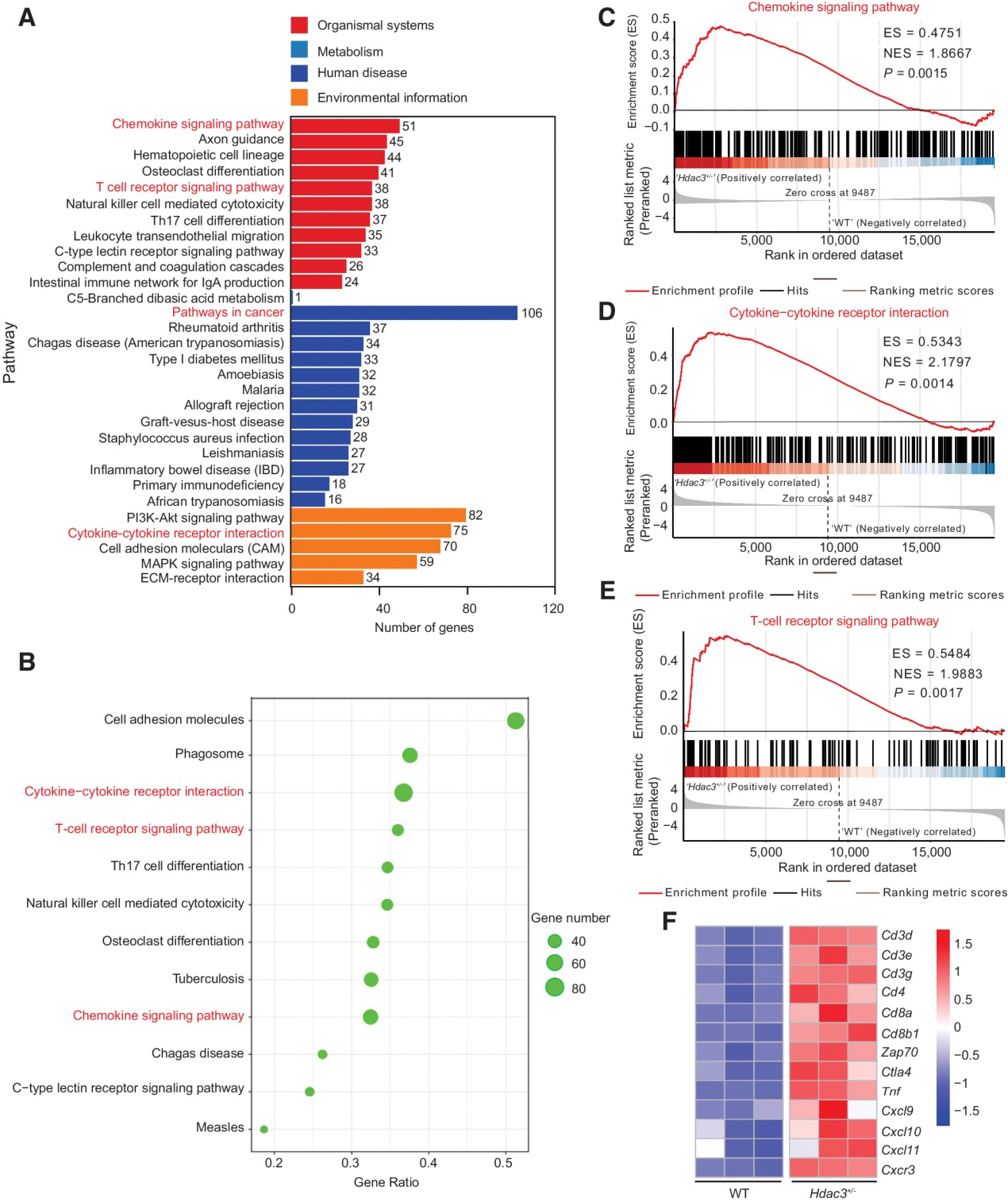
圖4:Hdac3缺乏上調趨化因子信號通路和TCR信號通路
(5) HDAC3下調IFN誘導的趨化因子Cxcl9, 10, 11的表達
趨化因子Cxcl9, 10, 11是IFN誘導基因,IFN信號通路在抗腫瘤免疫應答中起重要作用。因此,我們定量分析了在IFNα-,IFNβ-和IFNγ處理的WT和Hdac3?/? MCA205細胞中Cxcl9, 10, 11的mRNA的表達。qRT-PCR結果顯示,與WT型細胞相比,IFN處理的Hdac3?/?細胞中Cxcl9, 10, 11 mRNA表達水平明顯升高(圖5A)。接下來,我們通過ELISA檢測在IFNα-,IFNβ-和IFNγ處理的WT和Hdac3?/? MCA205細胞培養上清中CXCL9, 10, 11的濃度。與mRNA表達相似,與WT細胞相比,在Hdac3?/? MCA205細胞培養的上清中CXCL9, 10, 11蛋白濃度也更高(圖5B)。為了證實Cxcl9, 10, 11在體內Hdac3+/? MCA205腫瘤中表達上調,我們用ELISA法檢測了腫瘤組織中CXCL9, 10, 11的濃度,并發現CXCL9, 10, 11在Hdac3+/? MCA205腫瘤的表達也高于WT MCA205腫瘤(圖5C)。這些結果表明,Hdac3缺失上調了腫瘤細胞中Cxcl9, 10, 11的表達。
為了進一步證實HDAC3去乙酰化酶活性對Cxcl9, 10, 11表達的作用,我們在IFN處理的MCA205細胞中加入I類HDAC抑制劑Entinostat和HDAC3特異性抑制劑RGF966,通過qRT-PCR檢測Cxcl9, 10, 11 mRNA的表達。HDAC抑制劑Entinostat和RGF966也誘導Cxcl9, 10, 11 mRNA表達上調(圖5D)。相應地,我們也檢測到Cxcl9, 10, 11受體基因Cxcr3在Hdac3+/? MCA205腫瘤中表達上調(圖5E)。這些結果表明,HDAC3調節Cxcl9, 10, 11 mRNA的表達,并依賴于其去乙酰化酶的活性。
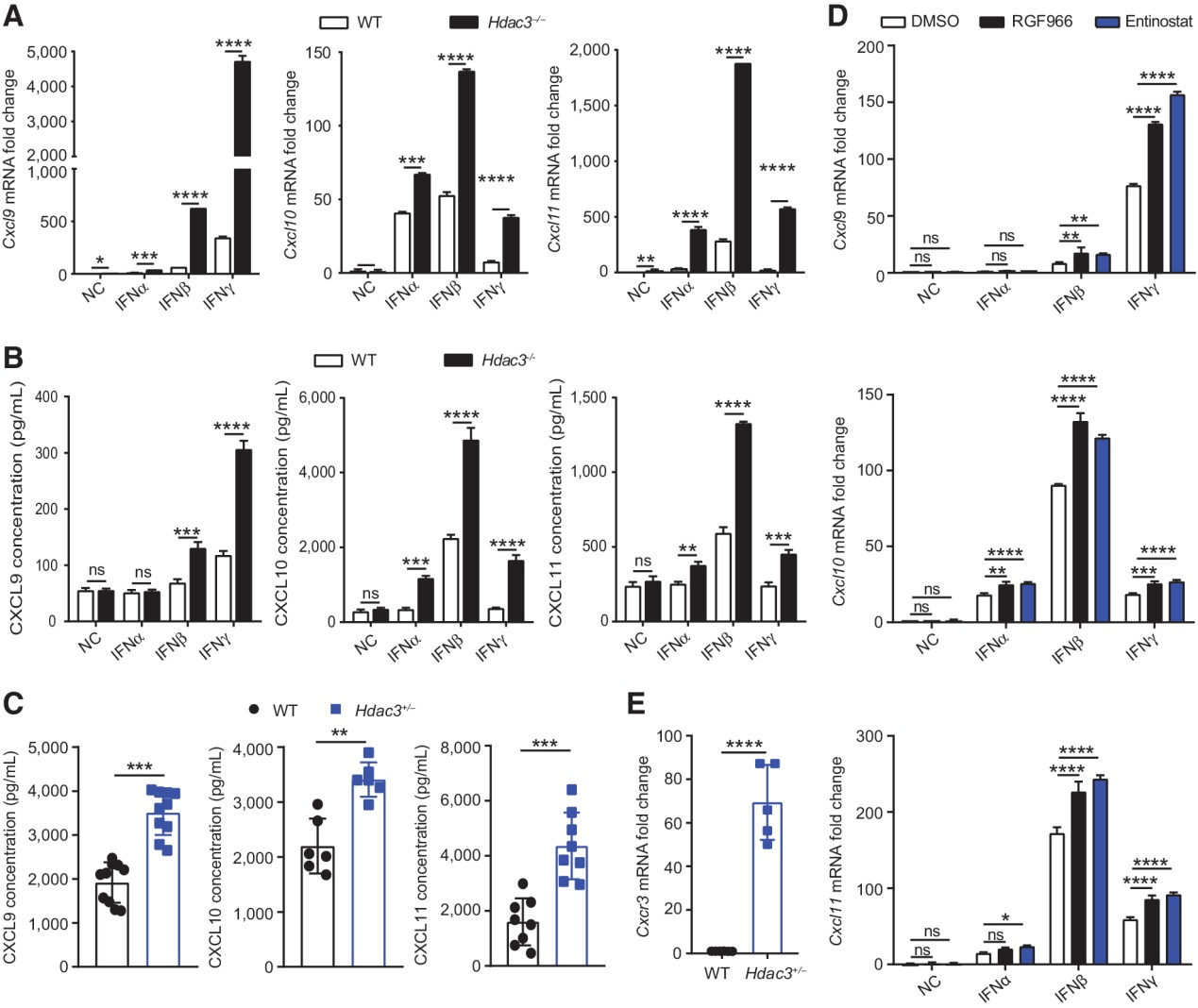
圖5:HDAC3下調IFN誘導的趨化因子Cxcl9, 10, 11的表達
(6) HDAC3直接結合并使Cxcl10基因啟動子去乙酰化
我們試圖探索HDAC3調控Cxcl9, 10, 11表達的機制。首先,我們比較了IFNα-、IFNβ-和IFNγ處理與未處理的MCA205細胞中Cxcl9, 10, 11 mRNA的相對表達。在MCA205細胞中,Cxcl10的表達高于Cxcl9和Cxcl11 (補充圖S6A和S6B),表明Cxcl10是MCA205腫瘤中負責CXCR3+ T細胞募集的主要趨化因子。HDAC3是一種表觀遺傳調節劑,可以介導組蛋白的去乙酰化修飾。接下來,我們通過ChIP-seq和ChIP-qPCR檢測了HDAC3和H3K27乙酰化(H3K27ac)在Cxcl10啟動子上的富集。在未處理和IFNβ刺激的WT MCA205細胞中,HDAC3在Cxcl10啟動子區域富集,H3K27ac水平較低。然而HDAC3+/?和HDAC3?/? MCA205細胞中Cxcl10啟動子上的HDAC3富集減少,IFNβ刺激后H3K27ac水平升高(圖6A-D)。這些結果表明HDAC3可以結合并抑制Cxcl10啟動子的H3K27ac。為了進一步確定HDAC3去乙酰化酶活性對Cxcl10轉錄的影響,我們用野生型HDAC3和去乙酰化失活突變體HDAC3?/? MCA205細胞(H134/135Q;補充圖S7A和S7B)。CCK-8 (補充圖S7C)、Ki67染色(補充圖S7D)和單克隆形成實驗(補充圖S7E和S7F)顯示,Hdac3過表達對MCA205細胞的活力和增殖沒有影響。此外,ChIP-qPCR和ChIP-seq結果顯示,WT HDAC3而不是HDAC3 H134/135Q突變體可以結合并抑制Cxcl10啟動子的乙酰化(圖6E-H)。進一步分析野生型和突變型HDAC3重組的HDAC3?/? MCA205細胞中Cxcl10 mRNA的表達,野生型HDAC3抑制Cxcl10 mRNA的表達,而H134/135Q-突變型HDAC3對Cxcl10 mRNA的表達無影響(圖6I)。此外,我們隨后將HDAC3?/? MCA205細胞接種到C57BL/6和裸鼠中,這些細胞由載體、野生型或H134/135Q突變型HDAC3重組。腫瘤生長監測顯示,在H134/135Q突變體中,而不是在H134/135Q突變體中,HDAC3挽救了腫瘤的生長(圖6J和K)。這些結果表明,HDAC3通過結合Cxcl10啟動子和去乙酰化附近的組蛋白來抑制Cxcl10基因的表達。
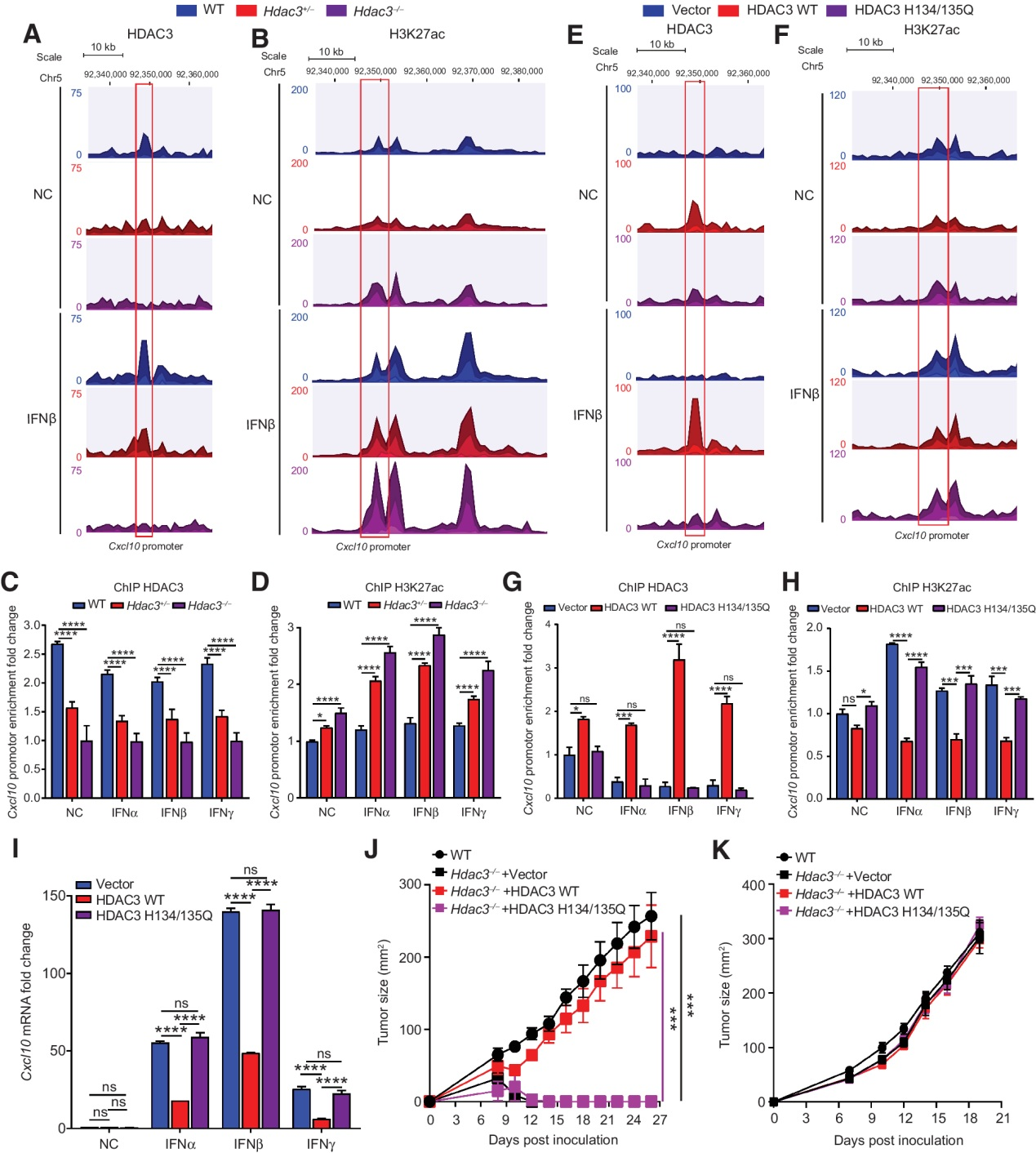
圖6:HDAC3直接結合并脫乙酰化Cxcl10基因啟動子
(7) CXCR3抗體阻斷以免疫依賴的方式拯救Hdac3缺陷的MCA205腫瘤生長
為了進一步驗證Hdac3缺乏促進CXCR3+ T細胞募集來介導抗腫瘤免疫應答的假設,我們使用CXCR3抗體在WT和HDAC3+/? MCA205腫瘤中阻斷CXCL9, 10, 11/CXCR3趨化因子信號通路。雖然TNFα抗體和IgG對HDAC3+/? MCA205腫瘤的緩慢生長無影響,但CXCR3抗體使HDAC3+/? MCA205腫瘤的生長恢復到與WT MCA205腫瘤相似的速度(圖7A)。通過流式細胞術進一步分析WT,IgG治療或CXCR3抗體治療的HDAC3+/? MCA205腫瘤的TILs,結果顯示,給予CXCR3抗體后,HDAC3+/? MCA205腫瘤中CD4+、CD8+、CXCR3+、CD4+CXCR3+和CD8+CXCR3+浸潤免疫細胞減少(圖7B)。免疫熒光染色結果顯示,與IgG組相比,CXCR3抗體組HDAC3+/? MCA205腫瘤中CD4+、CD8+和IFNγ+細胞數量減少(圖7C和D)。在Cxcr3抗體處理的HDAC3+/? MCA205腫瘤中,Cxcr3 mRNA也降低,這可能是由于表達Cxcr3的T細胞浸潤減少(圖7E)。這些結果表明,Hdac3缺陷腫瘤的生長降低可能是由于CXCR3+ T細胞浸潤到TME介導的抗腫瘤活性增強。總之,我們的研究表明,在TME中,HDAC3通過調節Cxcl9, 10, 11基因的表達和Cxcl9, 10, 11-CXCR3軸介導的T細胞募集參與抗腫瘤免疫,為HDAC3的抑制提供了重要的抗腫瘤機制(圖7F)。
我們還將MTX處理的WT、HDAC3+/?或HDAC3?/? MCA205細胞接種到左側攜帶WT MCA205腫瘤的小鼠右側,持續7天。我們發現,一側MTX處理的HDAC3+/?和HDAC3?/? MCA205細胞連接抑制了另一側WT MCA205腫瘤的腫瘤生長(補充圖S8A和S8B),這表明HDAC3缺陷的MCA205細胞在體內具有遠端抗腫瘤活性。為了確定CXCL10/CXCR3軸在HDAC3缺陷的MCA205細胞抗腫瘤活性中的作用,我們將WT, HDAC3+/?或HDAC3?/? MCA205纖維肉瘤細胞接種到左側WT MCA205腫瘤小鼠的右側,連續7天,然后靜脈注射CXCR3抗體或IgG對照。CXCR3抗體恢復了MCA205的腫瘤生長(補充圖S8C),提示Hdac3缺陷細胞的體內抗腫瘤活性依賴于CXCL10和CXCR3通路。

圖7:CXCR3抗體阻斷以免疫依賴的方式拯救Hdac3缺陷的MCA205腫瘤生長
(8) HDAC3與CXCL10在人HCC TME中的表達呈負相關
為了驗證HDAC3和CXCL10在人體樣本中的調節關系,我們收集HCC患者的惡性(n=86)和良性(n=41)組織,并進行TMAIHC評估HDAC3、CD8a、CXCL10和CXCR3的蛋白表達(補充表S1和S2)。癌組織中HDAC3的表達高于良性組織,而CXCL10的表達低于良性組織(補充圖S9A和S9B)。此外,通過Kaplan-Meier分析根據HDAC3和CXCL10水平分類的不同亞組的臨床結局,結果表明,HDAC3高表達或CXCL10低表達的HCC患者(cutoff=50%)的預后比HDAC3低表達或CXCL10高表達的HCC患者更差(補充圖S9C)。為了觀察HCC樣本中HDAC3、CXCL10、CD8a和CXCR3之間的關系,我們進一步分析了HDAC3、CD8a、CXCL10和CXCR3水平之間的相關性。HDAC3蛋白水平不高,但與CD8a和CXCL10的表達呈顯著負相關,CD8a的表達與CXCR3蛋白水平呈正相關(補充圖S9D)。這些臨床樣本的結果表明,HDAC3可能調節CXCL10的表達,進而影響CD8+ T細胞在HCC TME中的浸潤。
結論:我們證明了抑制HDAC3通過促進Cxcl9, 10, 11的表達來募集T細胞浸潤到TME中,從而抑制腫瘤生長。這項工作可能有助于指導未來的癌癥治療,通過在腫瘤細胞中使用HDAC3-特異性抑制劑來治療腫瘤細胞。
參考文獻:Li, L., Hao, S., Gao, M., Liu, J., Xu, X., Huang, J., Cheng, G., & Yang, H. (2023). HDAC3 Inhibition Promotes Antitumor Immunity by Enhancing CXCL10-Mediated Chemotaxis and Recruiting of Immune Cells. Cancer immunology research, 11(5), 657–673. https://doi.org/10.1158/2326-6066.CIR-22-0317.