






獨特的脂肪組織不變自然殺傷 T 細胞亞群控制小鼠脂肪細胞更新
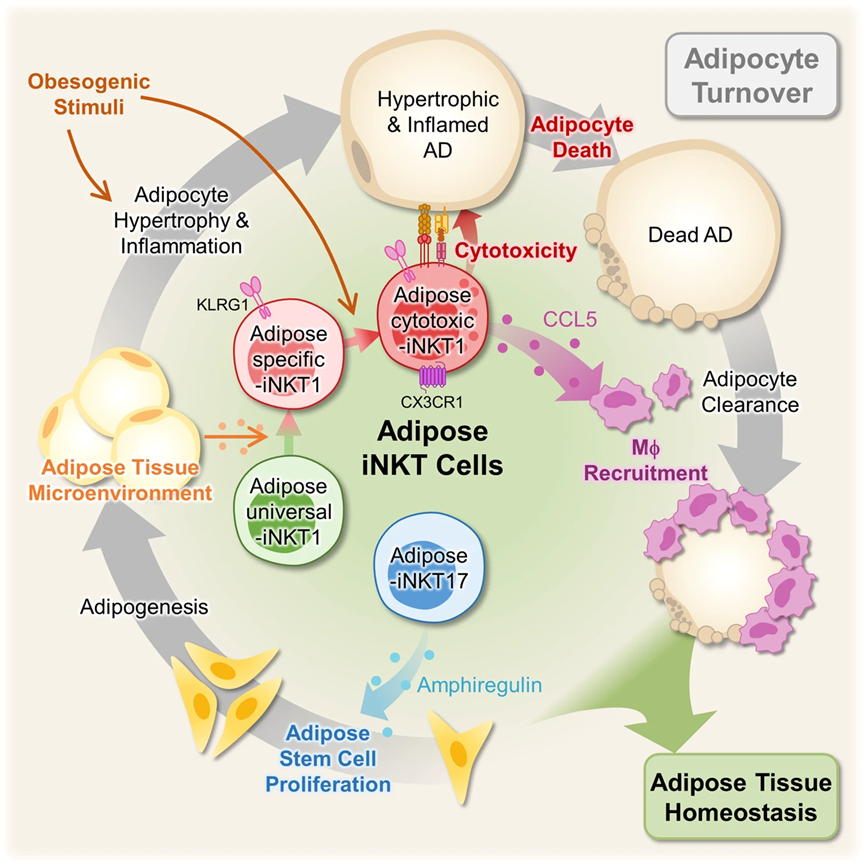
脂肪組織不可變自然殺傷T細胞(iNKT細胞)是肥胖動物脂肪組織穩態的關鍵細胞類型。然而,脂肪iNKT細胞的異質性及其在脂肪細胞周轉中的功能尚未徹底了解。在本研究中,我們利用單細胞RNA測序技術在瘦和肥胖小鼠中調查了脂肪iNKT細胞的轉錄異質性及其層次結構。我們報告了脂肪iNKT細胞的不同亞群通過調節脂肪組織的穩態影響脂肪細胞的死亡和出生。我們確定了KLRG1+ iNKT細胞作為脂肪組織中獨特的iNKT細胞亞群。通過移植實驗,我們發現KLRG1+ iNKT細胞在脂肪組織微環境中有選擇地生成,并在肥胖小鼠中分化為CX3CR1+細胞毒性亞群。此外,CX3CR1+ iNKT細胞特異性地殺死肥大和炎癥性的脂肪細胞,并通過CCL5招募巨噬細胞。此外,脂肪iNKT17細胞具有分泌AREG的潛力,而AREG參與刺激脂肪干細胞增殖。總體而言,我們的數據表明每個脂肪iNKT細胞亞群通過與脂肪細胞、脂肪干細胞和巨噬細胞的相互作用,在控制脂肪細胞周轉方面發揮關鍵作用。
該研究于2023年12月發表在《Nature communications》,IF:16.6。
機制圖:
技術路線:  結果:
結果:
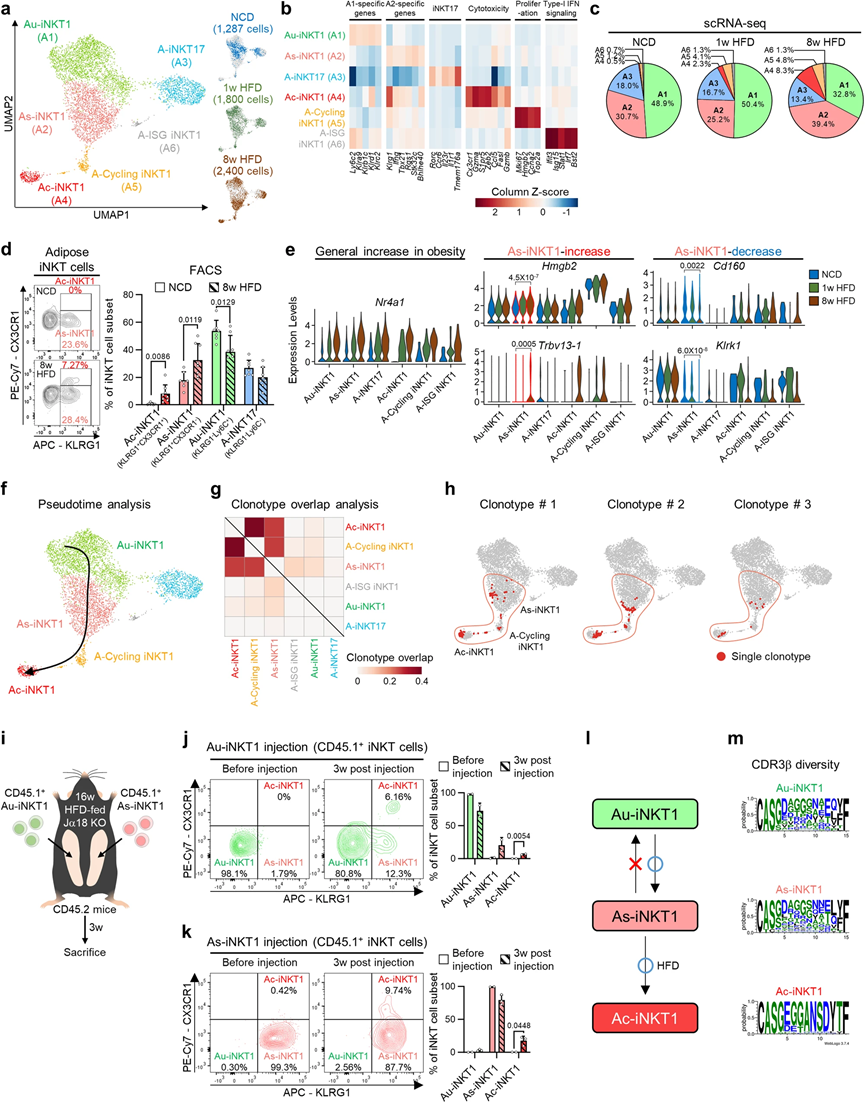
1、scRNA-seq 分析揭示脂肪 iNKT 細胞的不同亞群
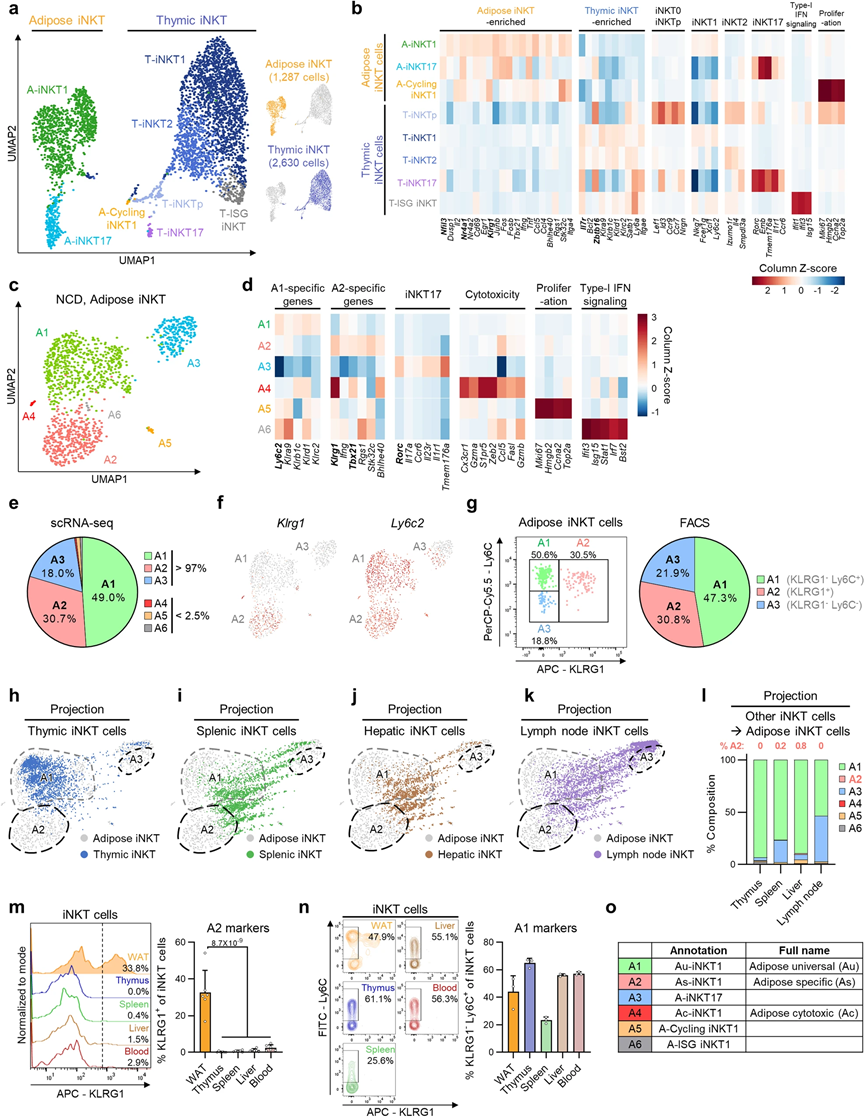
雖然與其他器官的iNKT細胞相比,脂肪iNKT細胞似乎具有組織特異性的特征,但目前很大程度上不清楚脂肪iNKT細胞的組織特異性特征是否有助于它們在脂肪細胞周轉中的功能。為了解決這個問題,我們對從附睪WAT中分選出的iNKT細胞進行了單細胞RNA測序,并將其與從胸腺分選出的iNKT細胞進行了比較,在胸腺中,iNKT細胞發育。如圖1a、b所示,脂肪iNKT細胞和胸腺iNKT細胞分別形成了不同的簇,表明它們在組織特異性基因表達方面存在明顯的分子特征:在WAT中為Nfil3(也稱為E4BP4)、Klrg1和Nr4a1,而在胸腺中為Zbtb16(也稱為PLZF)和Il7r。這些組織特異性基因表達譜與先前報道的比較脾和脂肪iNKT細胞的研究結果相似,暗示脂肪iNKT細胞中可能存在獨特的轉錄組譜。
在考察異質性方面,我們選擇了脂肪iNKT細胞并進行了重新聚類(圖1c)。在六個脂肪iNKT細胞亞群(A1–A6)中,大多數是表達Tbx21的iNKT1細胞(A1、A2和A4–A6)或表達Rorc的iNKT17細胞(A3)(圖1d)。除了A4和A6之外的大多數脂肪iNKT細胞亞群在轉錄上與先前報道的亞群相似。由于三個較小的亞群(A4–A6)僅占總脂肪iNKT細胞的2.5%以下(圖1e),我們主要關注進一步分析的三個主要亞群(A1–A3)。我們選擇KLRG1和Ly6C作為表面抗原,以區分這三個亞群(圖1f)。 KLRG1– Ly6C+、KLRG1+ 和 KLRG1– Ly6C– 的脂肪iNKT細胞分別與A1、A2和A3亞群相匹配。使用這些表面抗原區分脂肪iNKT細胞成功地反映了亞型比例和關鍵轉錄因子的表達(圖1g)。
盡管胸腺和脂肪iNKT細胞具有明顯的轉錄組譜,但它們組織特異性適應的起源目前很大程度上是未知的。為了表征脂肪iNKT細胞亞群的共有和組織特異性特征,我們進行了投影分析,使用了胸腺、脾、肝和淋巴結iNKT細胞。其他器官的iNKT細胞主要投影在A1亞群上,而不是A2亞群或A3亞群的下部分(圖1h–l)。這些數據暗示A2亞群和A3亞群的子集可能具有脂肪特異性特征,而A1亞群顯示出跨器官的共有基因表達譜。此外,脂肪iNKT細胞的組織特異性基因在A2亞群中高度表達(圖1b、d),表明A2亞群可能決定了脂肪iNKT細胞的組織特異性特征。
為了驗證A2是否確實是一個脂肪特異性亞群,對各種攜帶iNKT細胞的器官進行了測試,以確定它們是否表達A2亞群特異性的表面標記KLRG1。在其他器官中幾乎未檢測到KLRG1+ iNKT細胞,而來自WAT的iNKT細胞約有30%是KLRG1+的(圖1m)。為了測試這一模式是否與性別或品系有關,我們檢查了雌性C57BL/6和雄性BALB/c小鼠。由于它們表現出與雄性C57BL/6小鼠相似的模式,我們將A2亞群命名為‘脂肪特異性(As)-iNKT1’。此外,人類腹膜脂肪組織中也存在KLRG1+ iNKT細胞,表明人類可能含有類似的iNKT亞群。另一方面,由于其他器官中富含KLRG1– Ly6C+ iNKT細胞(圖1n),我們將A1亞群命名為‘脂肪通用(Au)-iNKT1’。A3–A6亞群分別命名為‘A-iNKT17’、‘脂肪細胞毒性(Ac)-iNKT1’、‘A-循環iNKT1’和‘A-干擾素誘導基因(ISG)-iNKT1’(圖1o),根據它們的特征基因表達譜(圖1d)。綜合這些數據,表明脂肪組織選擇性的KLRG1+ As-iNKT1細胞可能介導脂肪iNKT細胞的獨特特征。
2、脂肪組織微環境產生As-iNKT1細胞
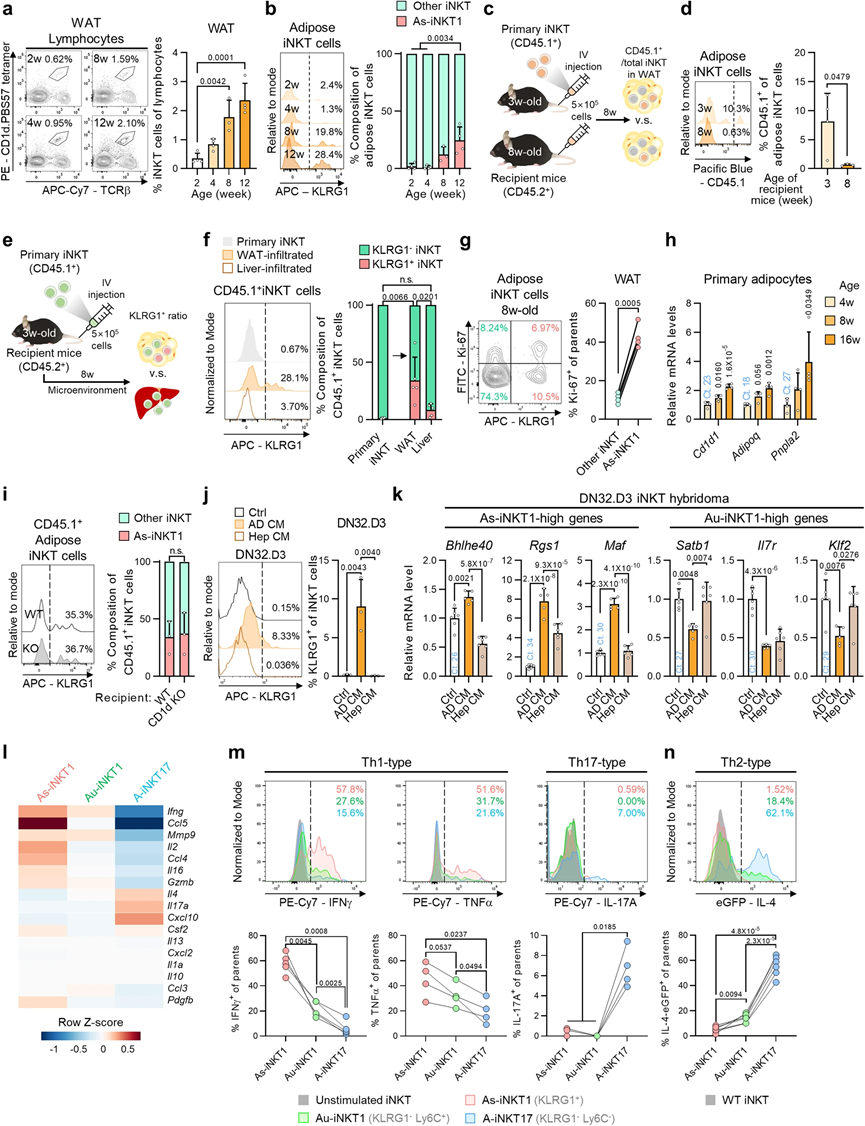
外周iNKT細胞在胸腺開始成熟,通過循環遷移到外周組織,并在外周器官中保持長期存在。與先前的報告一致,我們觀察到iNKT細胞在WAT中的數量隨年齡增加而增加(圖2a)。為了探究KLRG1+ As-iNKT1細胞在WAT中豐富的潛在機制,我們仔細研究了As-iNKT1細胞何時能夠出現在脂肪組織中。在4周之前,在WAT中只發現了極少數As-iNKT1細胞,而在8周后它們的比例增加(圖2b)。為了檢查4周后As-iNKT1細胞的生成情況,我們通過使用不同年齡的受體小鼠進行移植實驗,測試了iNKT細胞在發育過程中何時滲入脂肪組織(圖2c)。如圖2d所示,3周齡時注射的CD45.1+ iNKT細胞占總脂肪iNKT細胞的約8%,當它們在8周齡時注射時,這個比例大大減少。更重要的是,這些數據表明,大多數脂肪iNKT細胞在小鼠滿3周之前的早期階段就會滲入脂肪組織。因此,我們推測4周后As-iNKT1細胞的增加可能是由iNKT細胞所在的脂肪組織微環境介導的。為了測試這一點,我們從脾iNKT細胞建立的原代iNKT細胞被注射,然后根據浸潤的器官檢查KLRG1的表達程度(圖2e)。KLRG1+ iNKT細胞的比例在WAT中顯著更高,表明脂肪組織特異的微環境可能有助于KLRG1– iNKT細胞向KLRG1+ iNKT細胞的過渡(圖2f)。此外,注射的Au-iNKT1細胞的一個小亞群在三周后轉化為KLRG1+ As-iNKT1細胞。在WAT中,As-iNKT1細胞比其他iNKT細胞更具增殖能力,這可能促進它們在青春期后的增加(圖2g)。
接下來,我們鑒定了參與As-iNKT1細胞生成的脂肪組織特異性因子。為了找出4周后上調的脂肪組織特異性微環境因子,我們檢測了來自4、8和16周齡小鼠的脂肪細胞的mRNA表達譜。隨著年齡的增長,脂肪細胞上調了與微環境相關的基因,如Cd1d1、Adipoq和Pnpla2,分別對應于脂質抗原呈遞分子、脂聯素和脂解基因(圖2h)。為了檢查As-iNKT1細胞生成是否可能通過CD1d負載的脂質抗原(s)的慢性激活介導,我們將CD45.1+ iNKT細胞注射到年輕的WT小鼠或CD1d KO同胞中。如圖2i所示,在轉移的iNKT細胞中,As-iNKT1細胞的比例在兩個基因型之間沒有差異。然后,我們測試了脂肪細胞分泌的因子是否會影響As-iNKT1的生成。有趣的是,脂肪細胞培養基(AD CM)上調了As-iNKT1細胞的幾個標記基因,如Klrg1、Bhlhe40和Rgs1,同時下調了Au-iNKT1標記基因(圖2j、k)。這些模式在肝細胞培養基(Hep CM)處理組中不如在AD CM處理組中顯著(圖2j、k)。因此,這些數據表明,脂肪細胞分泌的因子可能在脂肪組織微環境中介導As-iNKT1細胞的生成。
在脂肪組織中,iNKT細胞活躍地分泌促炎和抗炎細胞因子來調節脂肪組織免疫。為了研究主要iNKT細胞亞群(包括As-iNKT1、Au-iNKT1和A-iNKT17細胞)的免疫調節特性,我們檢查了它們的細胞因子分泌模式。這些亞群通過激活后的KLRG1和Ly6C的表達模式進行區分。在三個主要亞群中,As-iNKT1細胞在激活后顯示出最高水平的IFNγ和TNFα產生(圖2l–n)。A-iNKT17細胞產生高水平的IL-17A和IL-4,而Au-iNKT1細胞顯示出中等水平的Th1和Th2型細胞因子產生(圖2l–n)。因此,脂肪iNKT細胞的炎癥調節特性似乎部分取決于各種代謝刺激下每個脂肪iNKT細胞亞群的相對比例,如1周高脂飲食喂養。
3、在肥胖癥中,As-iNKT1 細胞產生 Ac-iNKT1 細胞
在肥胖癥中,脂肪iNKT細胞通過清除有害的脂肪細胞,在維持脂肪組織穩態方面發揮了保護作用。盡管如此,脂肪iNKT細胞如何在肥胖癥中獲得或增強促進脂肪細胞清除的作用仍然不明確。為了研究肥胖引起的脂肪iNKT細胞的變化,我們對NCD、1周或8周高脂飲食喂養的同齡小鼠的脂肪iNKT細胞進行了scRNA-seq和TCR庫分析(圖3a)。即使在高脂飲食條件下,脂肪iNKT細胞仍然被分為與NCD條件相同的六個亞群(A1–A6),并且具有相似的標記基因表達(圖3b)。然而,在高脂飲食的影響下,iNKT細胞亞群的比例及其基因表達模式發生了改變。As-iNKT1、Ac-iNKT1和A-Cycling iNKT1細胞的比例在8周高脂飲食后增加,而Au-iNKT1細胞的比例相對減少,A-iNKT17和A-ISG iNKT1細胞的比例則沒有改變(圖3c)。A-Cycling iNKT1和A-ISG iNKT1細胞是少數亞群,占總脂肪iNKT細胞的不到5%(圖3c)。因此,我們決定在進一步的分析中排除A-Cycling iNKT1和A-ISG iNKT1細胞,主要集中在四個亞群上:Au-iNKT1(A1)、As-iNKT1(A2)、A-iNKT17(A3)和Ac-iNKT1細胞(A4)。為了將表達Klrg1的Ac-iNKT1細胞與As-iNKT1細胞區分開來,選擇CX3CR1作為Ac-iNKT1細胞的表面抗原。流式細胞分析證實了在8周高脂飲食后As-iNKT1和Ac-iNKT1細胞的增加,而Au-iNKT1細胞的減少(圖3d)。
為了了解肥胖如何調節As-iNKT1細胞的基因表達譜,分析了NCD和8周高脂飲食條件下的差異表達基因(DEGs)。大多數脂肪iNKT細胞亞群表現出與激活相關的表型,例如Nr4a1的上調(圖3e)。在8周高脂飲食后,As-iNKT1細胞上調了與增殖相關的基因(Hmgb2和Plk3),與Ifng表達相關的特征基因(Bhlhe40),以及特定的TCR Vβ鏈基因(Trbv13-1、Vβ8.3)(圖3e)。另一方面,As-iNKT1細胞中的一些NK受體基因,如Cd160和Klrk1,被下調(圖3e)。這些數據表明,在肥胖癥中,As-iNKT1細胞可能變得更具增殖能力,并且特定的As-iNKT1細胞可能選擇了特定的Vβ。其他脂肪iNKT細胞亞群在高脂飲食后也發生了變化。Au-iNKT1細胞上調了NK受體,如Klrd1和Klrc2,同時下調了Ifng。A-iNKT17細胞上調了Zbtb16和Rora,同時下調了一些與免疫相關蛋白(GIMAP)家族基因和MHC I分子有關的基因。
眾所周知,免疫細胞在病理刺激下可以分化為亞群。為了測試在肥胖癥中As-iNKT1細胞是否能夠分化為其他亞群,我們對三個iNKT1細胞亞群進行了偽時間分析:Au-iNKT1、As-iNKT1和Ac-iNKT1細胞。如圖3f所示,As-iNKT1細胞似乎是由Au-iNKT1細胞衍生出來的,并且可以分化為Ac-iNKT1細胞。克隆型重疊分析進一步驗證了Ac-iNKT1細胞與As-iNKT1細胞有相同的起源(圖3g,h)。此外,A-Cycling iNKT1細胞是As-iNKT1和Ac-iNKT1細胞的增殖亞群,這可能解釋了它們在肥胖癥中數量的增加(圖3g)。為了調查Au-iNKT1、As-iNKT1和Ac-iNKT1細胞在體內的層次關系,Au-iNKT1和As-iNKT1細胞分別注入了HFD喂養的iNKT細胞缺失的Jα18 KO小鼠的左右脂肪墊中(圖3i)。有趣的是,Au-iNKT1細胞分化為As-iNKT1和Ac-iNKT1細胞,而As-iNKT1細胞只分化為Ac-iNKT1細胞(圖3j–l)。此外,As-iNKT1細胞的分化伴隨著Ac-iNKT1細胞的CDR3β多樣性減少(圖3m),表明某些As-iNKT1細胞通過克隆擴張可能分化為Ac-iNKT1細胞。綜合這些數據,提出As-iNKT1細胞在暴露于肥胖刺激時可能會分化為Ac-iNKT1細胞。
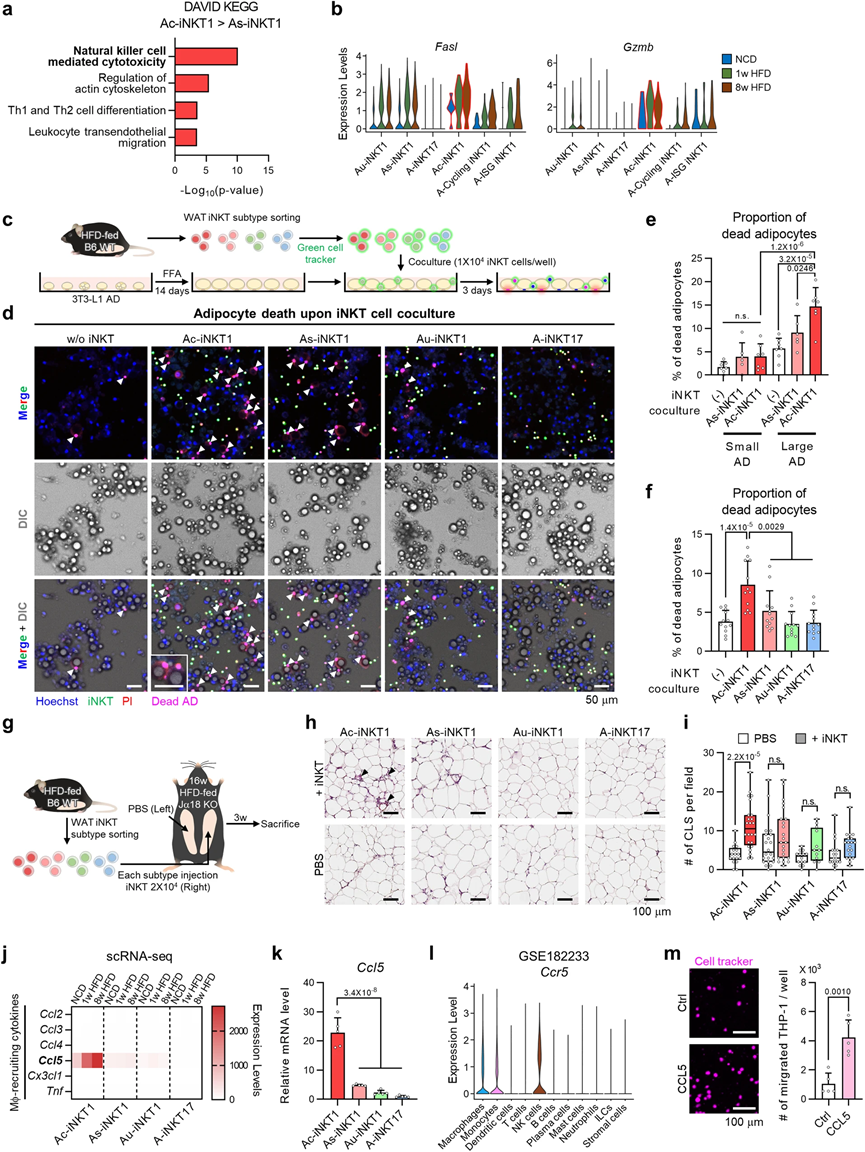
4、Ac-iNKT1 細胞通過分泌 CCL5 殺死肥大和炎癥脂肪細胞并招募巨噬細胞
最近,我們已經表明,脂肪iNKT細胞在高脂飲食條件下上調FasL,清除肥大和炎癥性的脂肪細胞。肥胖癥中Ac-iNKT1細胞數量增加和細胞毒基因表達的結果(圖3d,4a,b)促使我們檢查Ac-iNKT1細胞是否是能夠清除肥大和發炎的脂肪細胞的主要iNKT細胞亞群。為了解決脂肪iNKT細胞數量較少的技術問題,我們采用了使用α半乳糖二酰胺(α-GC)進行脂肪iNKT細胞的體內擴增,這是一種對iNKT細胞具有強大脂質抗原活性的物質。每個脂肪iNKT細胞亞群的基因表達譜和細胞因子產生在體內擴增后基本上沒有改變。首先,我們檢查了脂肪iNKT細胞亞群與脂肪細胞尺寸和死亡之間的關系。使用棕櫚酸(PA)或油酸(OA)過載的3T3-L1脂肪細胞(ADs),這被認為是具有促炎特性或具有無炎特性的肥大細胞,與每個iNKT細胞亞群進行共培養(圖4c)。有趣的是,Ac-iNKT1細胞選擇性地殺死大型ADs,而小型ADs不太容易被iNKT細胞清除(圖4d,e)。接下來,為了測試Ac-iNKT1細胞的細胞毒活性是否取決于ADs的炎癥特性,PA處理和OA處理的ADs,它們在炎癥特性上有所不同,被與每個iNKT細胞亞群共培養。如圖4f所示,AciNKT1細胞對PA處理的3T3L1 ADs(肥大和促炎的ADs)表現出最強的細胞毒性,而對OA處理的3T3-L1 ADs(肥大但不具有炎癥的ADs)則沒有顯示出顯著的細胞毒性。此外,CD1d中和明顯減少了Ac-iNKT1細胞引起的PA處理的AD死亡。這些數據表明,Ac-iNKT1細胞可能通過TCR/CD1d依賴的方式選擇性地清除具有肥大和促炎特性的脂肪細胞。為了在體內詳細研究Ac-iNKT1細胞的細胞毒性,我們將相同數量的脂肪iNKT細胞亞群注射到HFD喂養的iNKT細胞缺失的Jα18 KO小鼠的右脂肪墊中(圖4g)。在Ac-iNKT1細胞注射的脂肪墊中檢測到更多的冠狀狀結構(CLSs),這是死亡脂肪細胞的標志物(圖4h,i)。此外,Ac-iNKT1細胞顯示出與終末分化的CD8+效應T細胞(TE)相似的基因表達譜,表明Ac-iNKT1細胞在肥胖癥中將發揮關鍵作用,殺死肥大和發炎的脂肪細胞。
在死去的脂肪細胞周圍,巨噬細胞被招募來吞噬它們并形成CLS。為了測試Ac-iNKT1細胞是否能夠在清除死亡脂肪細胞后介導巨噬細胞的招募,檢查了與巨噬細胞招募有關的細胞因子。如圖4j,k所示,Ccl5是在巨噬細胞招募細胞因子中最高表達的,且僅在Ac-iNKT1細胞中表達。Ccr5是Ccl5的受體,僅在巨噬細胞、單核細胞和NK細胞中表達(圖4l),這表明CCL5可能通過招募巨噬細胞來清理死亡的脂肪細胞。一致性地,我們觀察到CCL5能夠介導THP1單核細胞的招募(圖4m)。綜合這些數據,清楚地表明Ac-iNKT1細胞可能通過CCL5選擇性地清除肥大和發炎的脂肪細胞,并通過招募巨噬細胞來清理死亡的脂肪細胞。
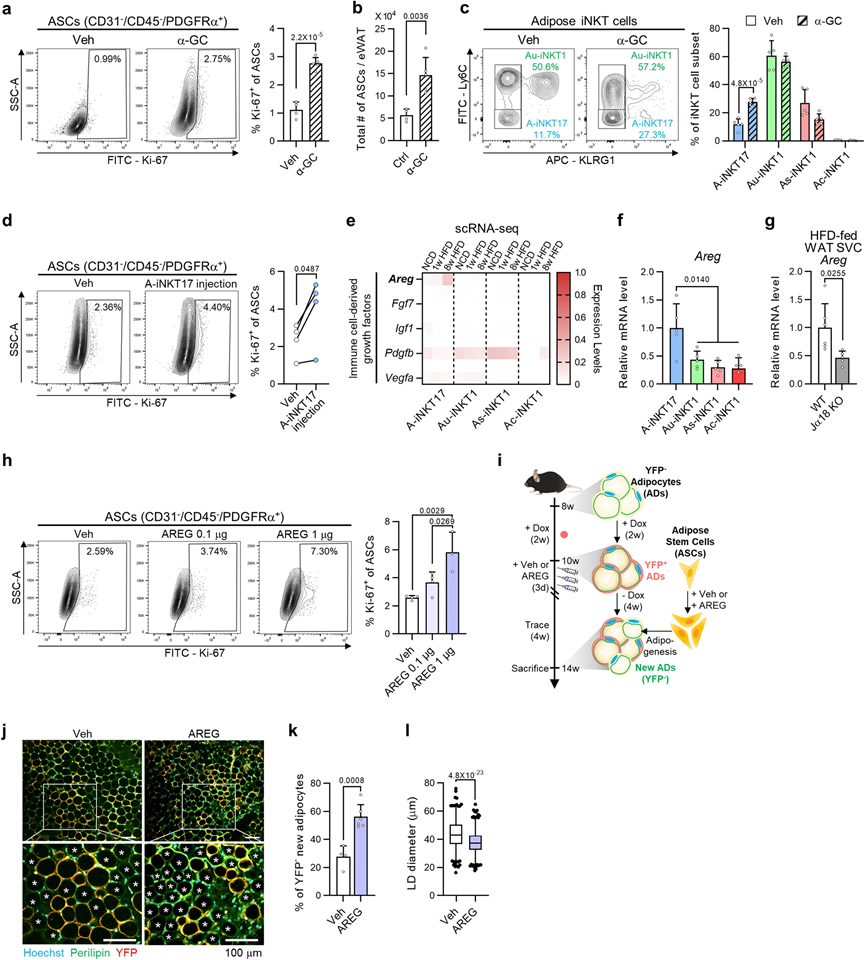
5、A-iNKT17細胞通過分泌雙調蛋白刺激脂肪干細胞增殖
目前尚不清楚脂肪iNKT細胞激活后ASC增殖是否歸因于死亡脂肪細胞釋放的因子,還是脂肪iNKT細胞與ASC直接相互作用的結果。為了驗證這一假設,我們向瘦小鼠注射α-GC。α-GC增加了脂肪組織中增殖ASC的比例,而沒有刺激脂肪細胞的凋亡(圖5a)。在Jα18 KO小鼠中,α-GC誘導的ASC增殖被iNKT細胞耗竭所廢除。此外,α-GC進一步增加了WAT中ASC的總數(圖5b)。這些數據表明,脂肪iNKT細胞在激活后可能直接刺激ASC增殖。
為了了解哪些脂肪iNKT細胞亞群將參與ASC增殖,我們采用了體內方法。由于α-GC注射顯著增加了A-iNKT17細胞的比例(圖5c),我們試圖驗證A-iNKT17細胞在ASC增殖中的作用。當將A-iNKT17細胞注射到HFD飼養的Jα18 KO小鼠的脂肪墊中時,注射A-iNKT17細胞增加了增殖ASC的比例,而其他脂肪iNKT細胞亞群的比例未發生改變(圖5d)。然后,為了確定A-iNKT17細胞中的哪種因子可能刺激ASC增殖,我們檢查了脂肪iNKT細胞亞群的基因表達譜。已經有報道表明免疫細胞通過生長因子或細胞因子與上皮或間質細胞進行活躍的交流,促進組織再生。有趣的是,在先前報道的免疫細胞衍生的生長因子中,在肥胖小鼠中A-iNKT17細胞中獨特地表達了刺激肌肉修復期間衛星細胞分化的amphiregulin(Areg),其表達水平逐漸增加(圖5e)。此外,Areg的mRNA水平在A-iNKT17細胞中似乎豐富,并在HFD飼養的iNKT細胞耗竭小鼠中下調(圖5f,g)。為了檢驗AREG是否能夠刺激ASC增殖,我們向瘦小鼠注射AREG。如圖5h所示,AREG以劑量依賴的方式增強了增殖ASC的比例。同時,通過抑制amphiregulin受體表皮生長因子受體(EGFR),抑制了αGC誘導的ASC增殖。接下來,為了調查AREG驅動的ASC增殖是否確實會導致脂肪細胞生成,我們采用了脂肪細胞譜系追蹤小鼠(adiponectinrtTA; TRE-Cre; rosa26-loxpstop-loxp-YFP)。通過青霉素的注射,現有的脂肪細胞被YFP標記,而載體或AREG注射后新形成的脂肪細胞則未被YFP標記(圖5i)。AREG注射增加了perilipin陽性且YFP陰性的新脂肪細胞的比例(圖5j,k),暗示AREG驅動的ASC增殖將提高脂肪生成的潛力。此外,AREG注射的WAT似乎顯示出較小的脂肪細胞大小(圖5l),這在激活的脂肪生成的脂肪組織中觀察到。鑒于相較于胸腺、脾和肝,WAT中iNKT17細胞的比例較高,似乎A-iNKT17-AREG軸在調節ASC增殖中發揮了重要作用。綜合而言,這些數據表明A-iNKT17細胞衍生的AREG可能是刺激ASC增殖和脂肪生成的關鍵介質之一,參與了脂肪組織的穩態。
實驗方法:
scRNA-seq、TCR分析、脂肪組織分離、FACS、共培養、免疫組織化學、THP-1單核細胞遷移測定、RT-qPCR。
參考文獻:
Han, S.M., Park, E.S., Park, J. et al. Unique adipose tissue invariant natural killer T cell subpopulations control adipocyte turnover in mice. Nat Commun 14, 8512 (2023).