






METTL3通過激活JAK1/STAT3信號通路促進結直腸癌進展
METTL3介導的N6-甲基腺苷(m6A)修飾在多種癌癥中的作用已被闡明,但其在結直腸癌中發揮作用的具體機制仍不清楚。本研究中,作者發現在結直腸癌中,上調的甲基轉移酶樣3(METTL3)在基因調控中發揮依賴于甲基轉移酶活性和不依賴于甲基轉移酶活性的兩種功能。JAK1和STAT3的增加共同促進p-STAT3信號通路的激活,并進一步上調包括VEGFA和CCND1在內的下游效應物的表達,最終導致體外和體內癌細胞增殖和轉移的增強。機制上, METTL3將m6A沉積在JAK1轉錄本的3'非翻譯區,依靠YTHDF1的識別促進JAK1的翻譯。此外,METTL3被重新分配到STAT3啟動子上,并與NF-κB協同促進STAT3的轉錄,而這是獨立于METTL3甲基轉移酶活性實現的。總之,該研究揭示了METTL3作為m6A writer和轉錄調控因子的雙重作用,它們在同一信號通路中共同作用,驅動結直腸癌惡性發展。該研究于2023年11月發表在《Cell Death & Disease》,IF:9.0。
技術路線:

主要研究結果:
1. m6A和METTL3在結直腸癌中上調
為探究m6A機制在結直腸癌中的作用,研究人員利用Northern點印跡技術檢測10個配對的癌癥組織和正常組織切片中的總RNA m6A水平。軟件分析表明,與正常組織相比,癌癥組織中的 m6A 水平呈上調趨勢,這表明 m6A 在結直腸癌的發展過程中起著協同促進作用(圖 1A)。隨后,利用生物信息學方法篩選人大腸癌中的甲基轉移酶和去甲基化酶(METTL3/14、WTAP、FTO和ALKBH5)的表達情況,發現只有甲基轉移酶的催化核心METTL3在癌組織中較正常組織有顯著上調(圖1B)。更重要的是,轉移灶的METTL3表達高于原發腫瘤(圖 1C),凸顯METTL3與惡性腫瘤的關系。然后,通過Western印跡和免疫組化染色評估METTL3在臨床樣本中的蛋白水平表達(圖1D、E)。與mRNA分析結論一致,與配對的正常組織相比,結直腸癌和轉移樣本中的METTL3蛋白水平明顯升高。此外,與正常人結腸上皮細胞(FHC)相比,所有檢測到的人結直腸癌細胞系都顯示出METTL3表達增加和 m6A 修飾水平上調(圖 1F,G)。上述數據表明,METTL3 和 m6A 的上調與結直腸癌的惡性程度和進展有關。
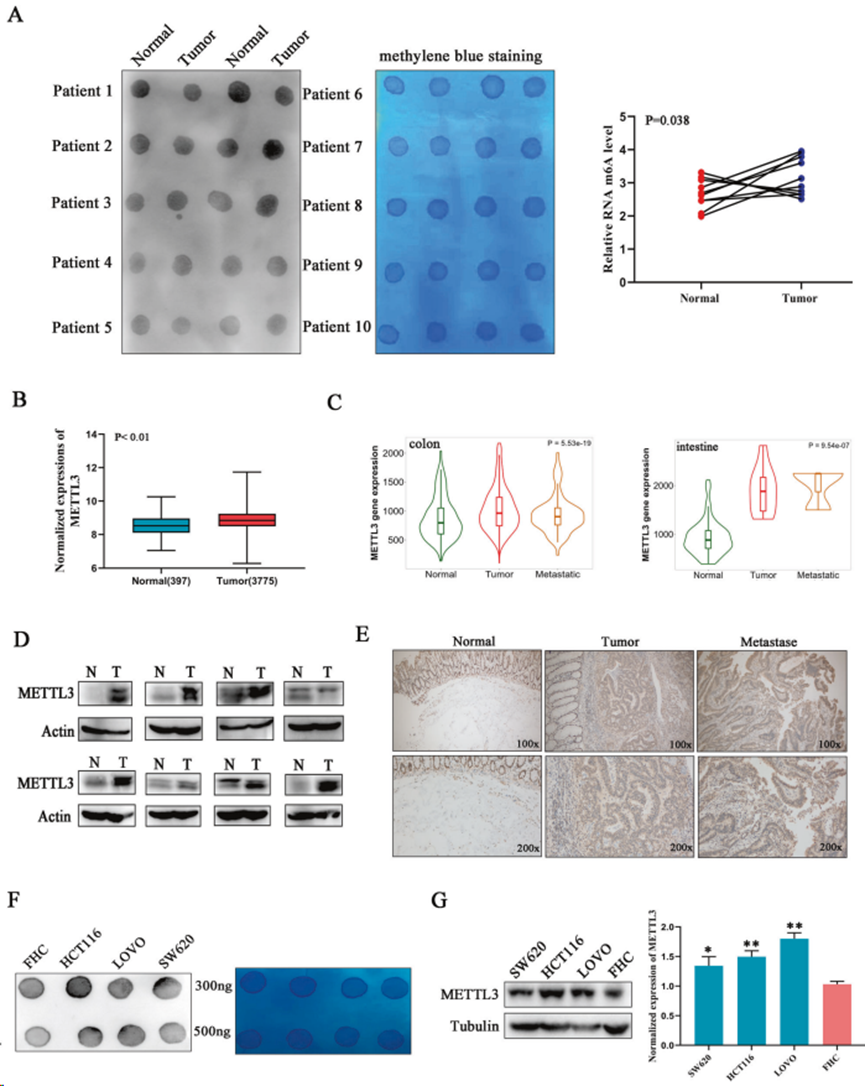
圖1. m6A和METTL3在結直腸癌中上調
2. METTL3在結直腸癌細胞中的協同促進作用
為進一步闡明METTL3介導的m6A靶向基因調控機制,利用慢病毒介導的shRNA轉染技術建立METTL3穩定下調的結直腸癌細胞。在HCT116和LoVo 細胞系中,METTL3 的敲除效率分別通過實時定量 PCR 和 Western 印跡得到證實(圖 2A,B)。進行細胞增殖、集落形成和MTT試驗。與對照組相比,METTL3 減少的癌細胞對細胞增殖和集落形成有明顯的抑制作用(圖 2C,D)。Transwell顯示,METTL3 基因敲除顯著削弱 HCT116 和 LoVo 細胞的遷移能力(圖 2E)。細胞增殖抑制通常是由細胞周期停滯或細胞死亡引起的。因此,通過流式細胞術檢測癌細胞的細胞周期和細胞凋亡。與對照組相比,METTL3沉默的細胞中有更多的細胞被抑制在G1/G0期,停留在G2/M期的細胞比例明顯下降(圖2F),但細胞凋亡在這兩組之間沒有觀察到明顯的差異(圖2G)。因此,METTL3可能通過調節細胞周期和遷移而發揮共促進作用,但并不依賴于細胞凋亡。
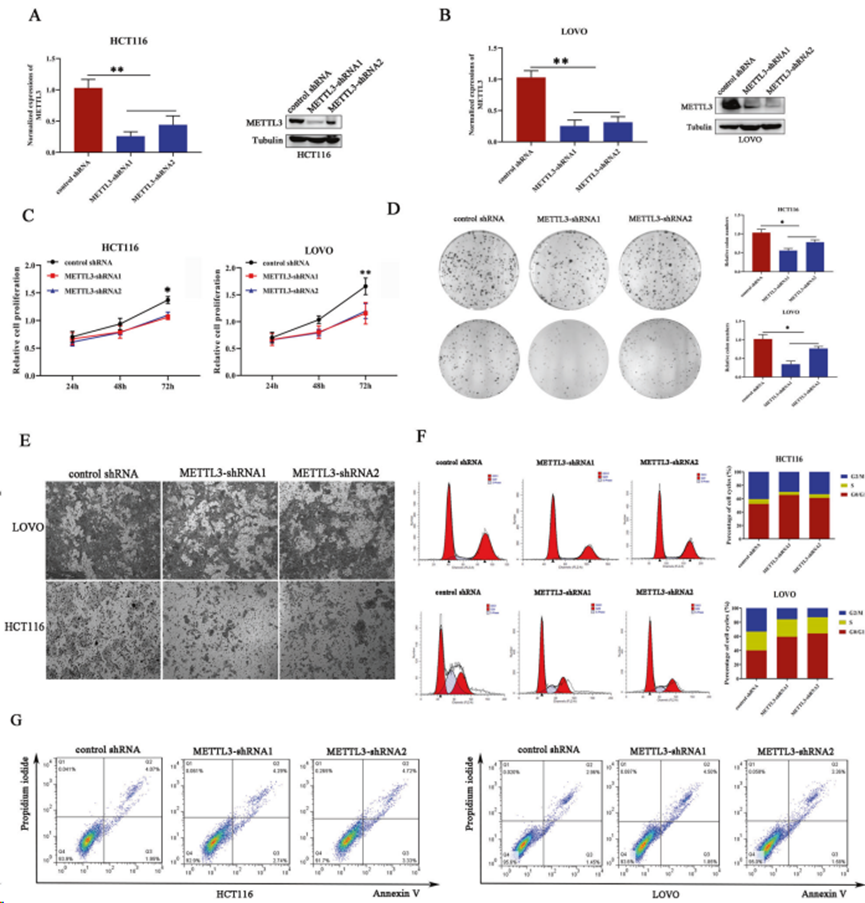
圖2. METTL3在結直腸癌細胞中的協同促進作用
3. 沉默METTL3對JAK1/STAT3信號通路的影響
考慮到METTL3對RNA修飾的作用,研究人員通過點印跡法檢測METTL3 缺失的癌細胞中RNA m6A的水平。與對照組相比,沉默METTL3后,癌細胞中的m6A水平明顯下降(圖 3A),這與之前的報道一致。為解讀METTL3對m6A 靶基因表達的作用,研究人員對敲除METTL3的HCT116細胞進行甲基化RNA 免疫沉淀和RNA測序(MeRIP-seq)。測序結果顯示,大部分m6A峰分布在mRNA 3'UTR和翻譯停止點附近的位點,m6A 沉積量最高(圖 3B),這與之前的測序結果一致。挑出m6A峰值發生變化(log2 ≥ 1)的基因進行通路富集統計。其中,JAK/STAT 信號通路的出現引起注意(圖3C)。對JAK1/STAT3信號通路的狀態進行研究,Western印跡結果顯示,沉默METTL3顯著抑制STAT3在Tyr705 殘基上的磷酸化。令人驚訝的是,在METTL3敲除的癌細胞中也觀察到JAK1和STAT3表達的明顯下降,而對照組則沒有(圖3D)。為明確METTL3調控JAK1和STAT3的表達是否依賴于其甲基轉移酶活性,使用m6A抗體沉淀含m6A修飾的RNA,并對其進行了定量實時PCR檢測。為排除脫靶的可能性,設計兩種針對 STAT3 不同位點的成對引物。實時 PCR 結果顯示,敲除METTL3后,帶有m6A修飾的JAK1 mRNA量減少,但帶有m6A修飾的STAT3 mRNA量沒有差異(圖3E),表明 METTL3 可能以一種與 m6A 無關的方式介導 STAT3 的表達。
隨后,利用CRISPR-Cas9技術在HCT116中構建METTL3基因敲除細胞。經過單細胞選擇后,用 Western blot 檢測十個細胞的結腸,發現只有結腸-4(KO-C4)和結腸-7(KO-C7)顯示出 METTL3 基因敲除效率(圖3F)。對于 JAK1/STAT3通路分析,METTL3 基因敲除細胞的Western印跡檢測結果與 METTL3 基因敲除細胞的結果相同(圖3G)。實時 PCR 檢測發現,METTL3 基因敲除在 mRNA 水平上顯著下調STAT3的表達,但對JAK1 mRNA的表達沒有影響(圖 3H)。為進一步驗證 METTL3 介導的對 JAK1 和 STAT3 的調控并非脫靶效應,在METTL3 基因敲除細胞中外源過表達野生型METTL3(WT-METTL3)和失去甲基轉移酶活性的突變型 METTL3(D395A/W398A)。發現,無論是WT-METTL3還是突變體METTL3的過表達都能挽救STAT3的表達,但只有WT-METTL3能挽救JAK1的表達,突變體METTL3的過表達對JAK1的影響減弱(圖 3I)。上述數據表明,METTL3以m6A依賴的方式在蛋白水平上調控JAK1的表達,而不是在mRNA水平上,并影響STAT3 mRNA的表達,從而共同促進結直腸癌細胞中JAK1/STAT3信號通路的激活。
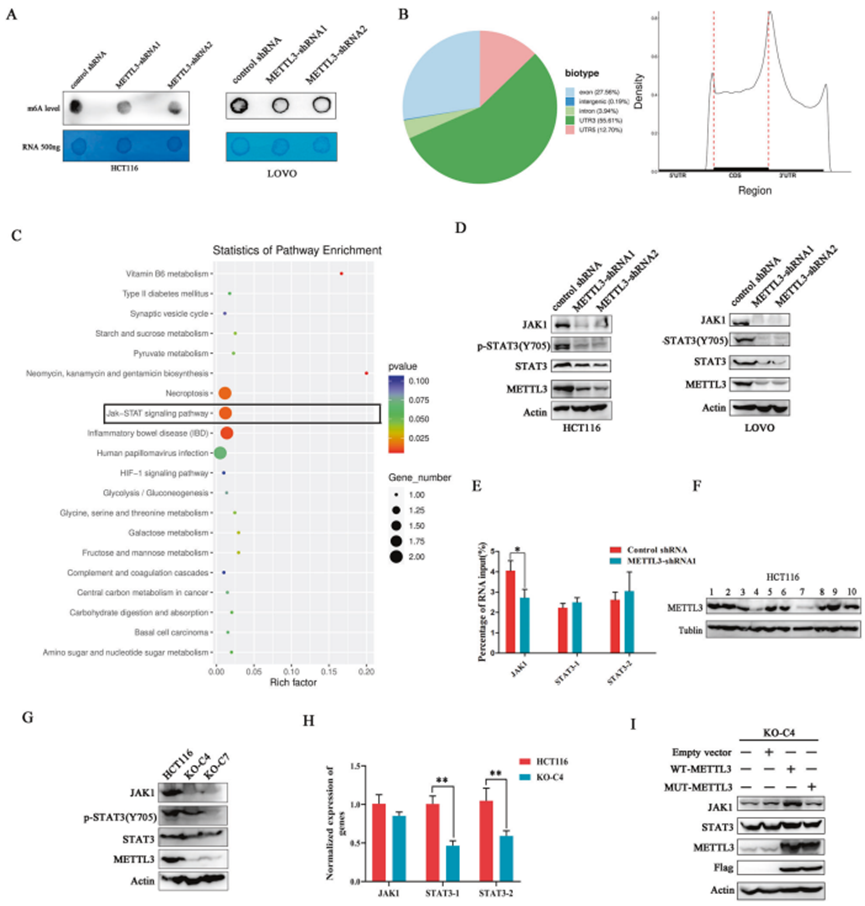
圖3. 沉默METTL3對JAK1/STAT3信號通路的影響
4. YTHDF1以m6A依賴性方式促進JAK1 mRNA翻譯
使用蛋白酶抑制劑 MG132 來阻斷蛋白酶體介導的蛋白降解。發現,MG132 處理未能補充METTL3敲除介導的JAK1下調(圖4A),表明METTL3可能參與JAK1翻譯。在 HCT116 細胞系中構建穩定沉默YTHDF1的細胞(圖4B)。Western印跡檢測顯示,YTHDF1敲除能有效抑制STAT3磷酸化,降低JAK1的表達水平,但對STAT3的表達沒有影響(圖4C)。進行RNA 相關免疫沉淀(RIP)和實時定量 PCR(圖 4D),發現JAK1 mRNA富集在YTHDF1免疫沉淀物中,METTL3基因敲除后,這種相互作用明顯減弱(圖4E,F),突出m6A在YTHDF1和JAK1 mRNA相互作用中的不可或缺性。通過掃描HCT116中的MeRIP-seq數據,發現JAK1 mRNA 3'UTR中分布著大量置信度極高的m6A峰,并預測出4個潛在的m6A位點(圖4G)。將 JAK1 3'UTR 序列克隆到雙熒光素酶 pmirGLO 載體中,并分別構建潛在 m6A 位點發生單堿基替換的突變載體(圖4H)。熒光素酶檢測結果表明,與野生型相比,轉染位點3和位點4存在m6A突變的載體的細胞,其熒光素酶活性明顯減弱,這意味著位于JAK1 mRNA 3'UTR位點3和位點4的m6A修飾是m6A介導的JAK1轉錄后調控的原因(圖4I)。此外,與對照組相比,YTHDF1的缺失也導致熒光明顯下降(圖4I),這進一步驗證YTHDF1在識別JAK1 m6A位點中的重要作用。上述數據表明,METTL3對JAK1表達的調控功能是通過YTHDF1識別m6A位點來增強靶基因翻譯的能力來實現的。
然后,研究YTHDF1在結直腸癌細胞中的作用。沉默YTHDF1明顯抑制細胞增殖和集落形成(圖 4J,K),與對照細胞相比,YTHDF1 缺失細胞的遷移能力也受到影響(圖4L)。此外,流式細胞術分析表明,停留在M期的細胞比例下降,更多的細胞停滯在S期(圖4M),這與在METTL3沉默細胞中觀察到的結果一致。綜合上述數據,METTL3依賴YTHDF1與JAK1 m6A位點的結合能力促進JAK1翻譯,導致STAT3通路激活和癌癥進展。
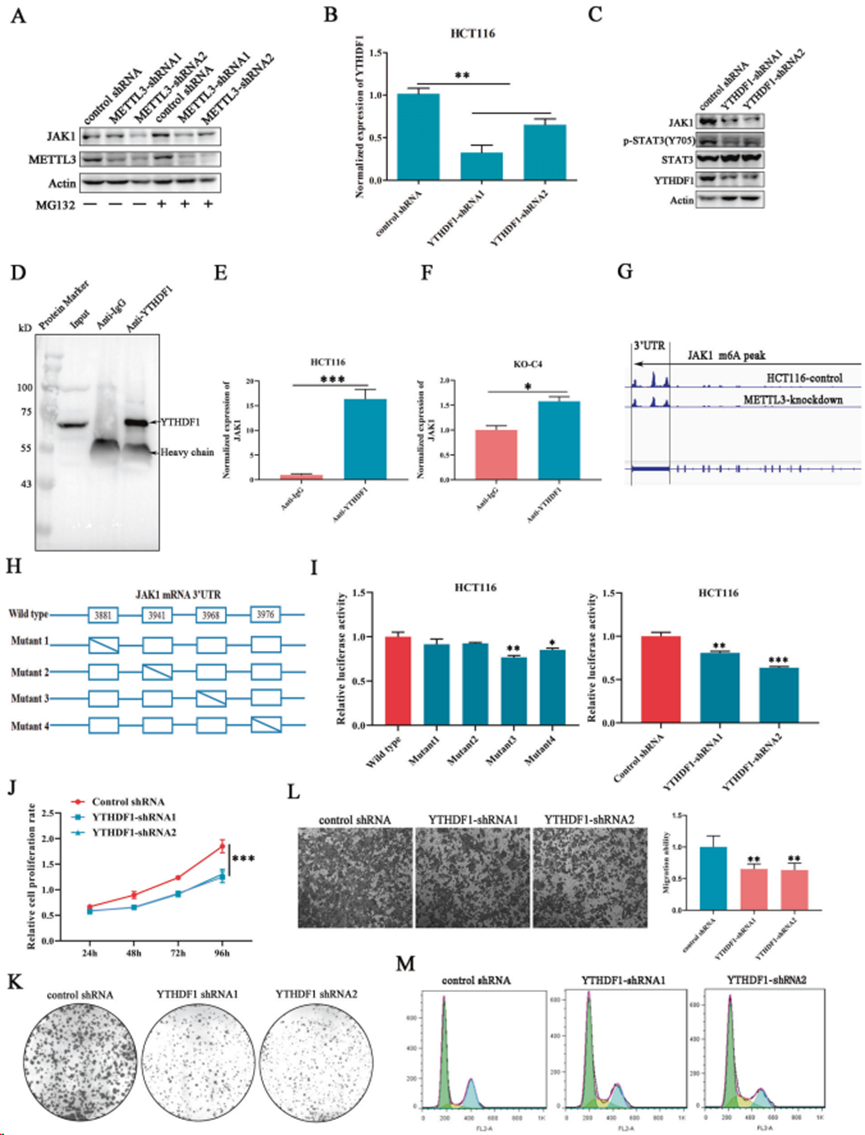
圖4. YTHDF1以m6A依賴性方式促進JAK1 mRNA翻譯
5. METTL3和NF-κB共同調控STAT3轉錄
使用放線菌素D中斷細胞的RNA合成。實時PCR檢測表明,在放線菌素 D暴露下,METTL3基因敲除組與對照組STAT3 mRNA降解率無明顯差異(圖 5A),說明STAT3 mRNA 降解不受METTL3影響。利用免疫熒光檢測METTL3 在HCT116細胞中的位置,發現METTL3大部分定位于細胞核中,在細胞質中幾乎觀察不到任何信號(圖5B)。隨后,參考已發表的METTL3和METTL14的ChIP-seq數據(GSE141992)進行生物信息學分析,發現METTL3結合位點在STAT3啟動子特異性地靠近轉錄起始點附近非常豐富(圖5C)。令人驚訝的是,在 STAT3 啟動子區域也發現NF-κB 的足跡(圖5C),這表明METTL3可能與 NF-κB協同調控STAT3的轉錄。隨后,在HCT116 細胞中分別對METTL3和 NF-κB進行ChIP 檢測。分離 PCR 產物,發現STAT3啟動子片段出現在 METTL3和NF-κB沉積物中,而抗IgG組中沒有(圖5D,E),這表明 METTL3/NF-κB與STAT3啟動子直接相互作用。
隨后,通過共免疫沉淀也證實METTL3與NF-κB之間的直接相互作用(圖5F)。為進一步探討NF-κB是否具有調控 STAT3 表達的能力,用siRNA抑制癌細胞中NF-κB 的表達,發現 STAT3 的表達適度下降(圖 5H)。此外,在線數據挖掘顯示,在臨床病理結直腸癌樣本中,NF-κB和STAT3的表達呈正相關(圖5G),這為 NF-κB在調控 STAT3 表達中的作用提供新的證據。最后,將包含轉錄起始位點上游450 bp和下游40 bp的序列構建成啟動子活性檢測質粒(圖5I)。此外,構建已知NF-κB結合位點突變的相同序列,并轉染到癌細胞中進行啟動子活性檢測。與野生型組相比,NF-κB突變明顯降低STAT3啟動子的活性,而且這種抑制作用在METTL3基因敲除的癌細胞中進一步擴大(圖5I)。上述數據共同表明,METTL3和NF-κB協同促進STAT3的轉錄。
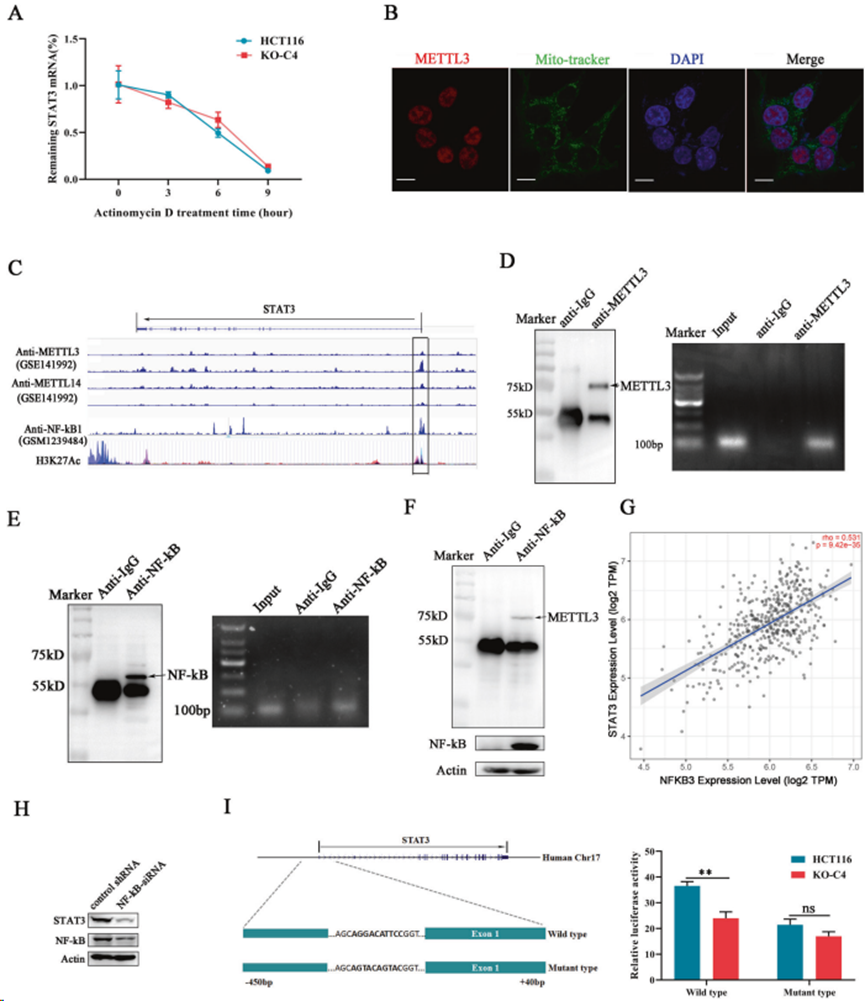
圖5. METTL3和NF-κB共同調控STAT3的轉錄
6. p-STAT3 信號失調的下游效應
對METTL3沉默的癌細胞中VEGFA的表達情況進行調查。METTL3的缺失在mRNA水平和蛋白水平上都明顯抑制了VEGFA 的表達(圖 6A,B)。根據研究結果,METTL3沉默通過誘導細胞周期停滯來抑制細胞增殖。在METTL3 缺失的細胞中,CCND1 的表達明顯下降,同樣的結果也在YTHDF1沉默的癌細胞中得到證實(圖 6C,D)。這些數據表明,METTL3可能通過激活JAK1/STAT3信號通路來調控VEGFA和CCND1的表達。使用 STAT3小分子抑制劑Stattic 阻斷HCT116癌細胞中STAT3的磷酸化。Western印跡顯示,STAT3的活性受stattic的抑制呈劑量依賴性,其下游因子包括VEGFA和CCND1在stattic處理后也出現下調(圖6E)。此外,stattic劑量依賴性地影響HCT116癌細胞的集落形成和遷移(圖6F,G)。數據庫分析顯示,與正常人群相比,VEGFA、CCND1 和 YTHDF1在結直腸癌組織中的表達均上調(圖6H)。在幾乎所有分析的癌癥類型中,METTL3與其靶標(包括JAK1、VEGFA、CCND1和STAT3)之間的表達相關性均為陽性(圖6I)。總之,上述數據揭示STAT3 信號通路在METTL3介導的結直腸癌細胞增殖和遷移中發揮重要作用。
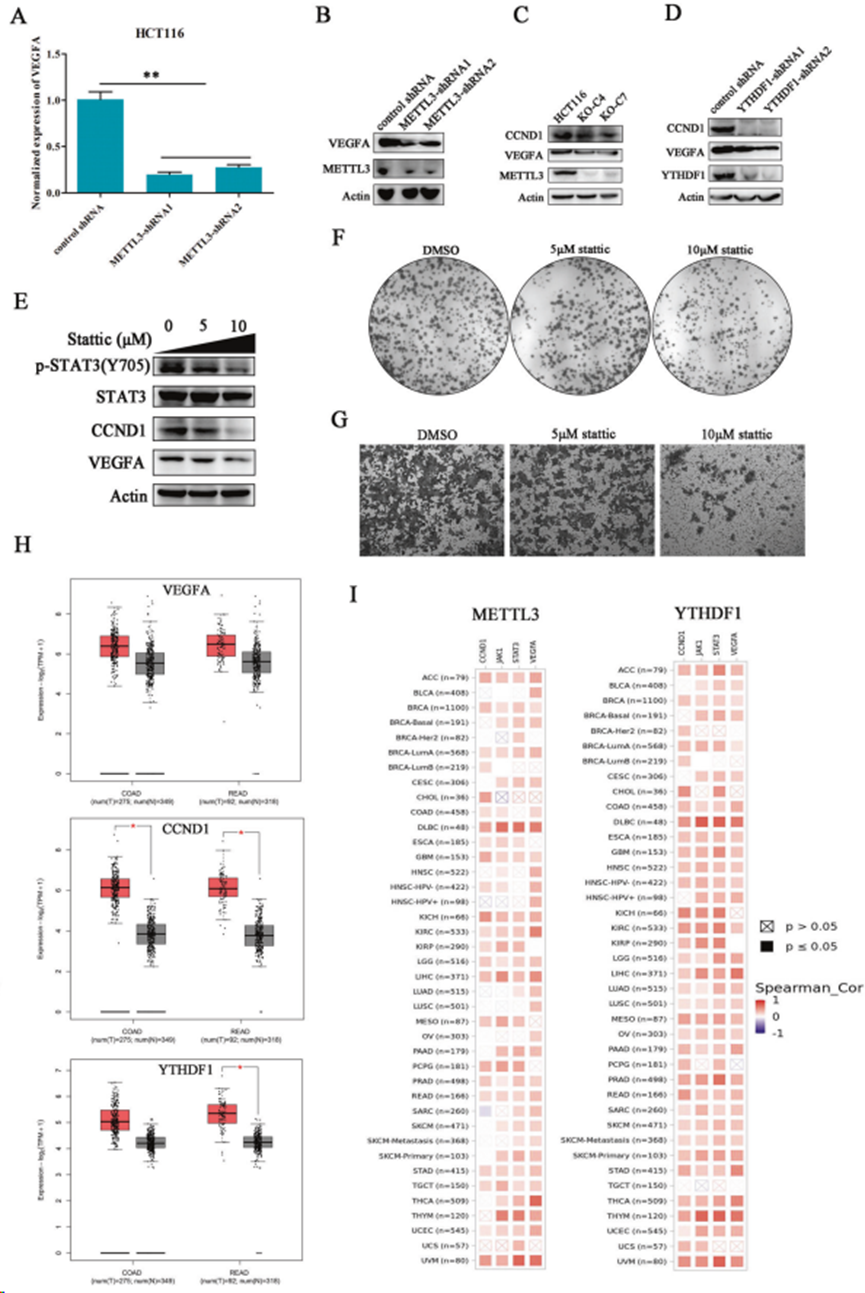
圖6. JAK1/STAT3 信號失調的下游效應
7. METTL3和YTHDF1促進體內腫瘤生長和轉移
將缺失或未缺失METTL3的結直腸癌細胞皮下注射到 BALB/c裸鼠體內。METTL3基因敲除有效延緩腫瘤生長(圖7A),因為與對照組相比,METTL3 基因缺失組的異種移植物的重量和大小均顯著減少(圖7B、C)。隨后,建立肺轉移小鼠模型,尾靜脈注射HCT116對照組細胞會導致嚴重的肺轉移,而缺失METTL3則幾乎完全消除轉移結節的形成(圖 7D,E)。所有這些都證實 METTL3通過促進細胞增殖和轉移在結直腸癌發生中的致癌作用。
將YTHDF1缺失的癌細胞注射到裸鼠體內,建立腫瘤小鼠和肺轉移小鼠模型。YTHDF1沉默后,腫瘤負荷明顯減輕(圖7F-H),與對照組相比,注射YTHDF1缺失細胞的小鼠肺組織切片中出現的轉移結節更少(圖 7I,J)。此外,STAT3 信號相關蛋白的表達水平在METTL3或YTHDF1缺乏的異種移植物中有所降低(圖7K),這與在細胞中觀察到的結果一致。總之,研究結果表明METTL3 以依賴和不依賴 m6A 的方式同時上調JAK1和STAT3的表達,從而促進STAT3 信號的激活,導致結直腸癌在體外和體內的增殖和進展(圖8)。
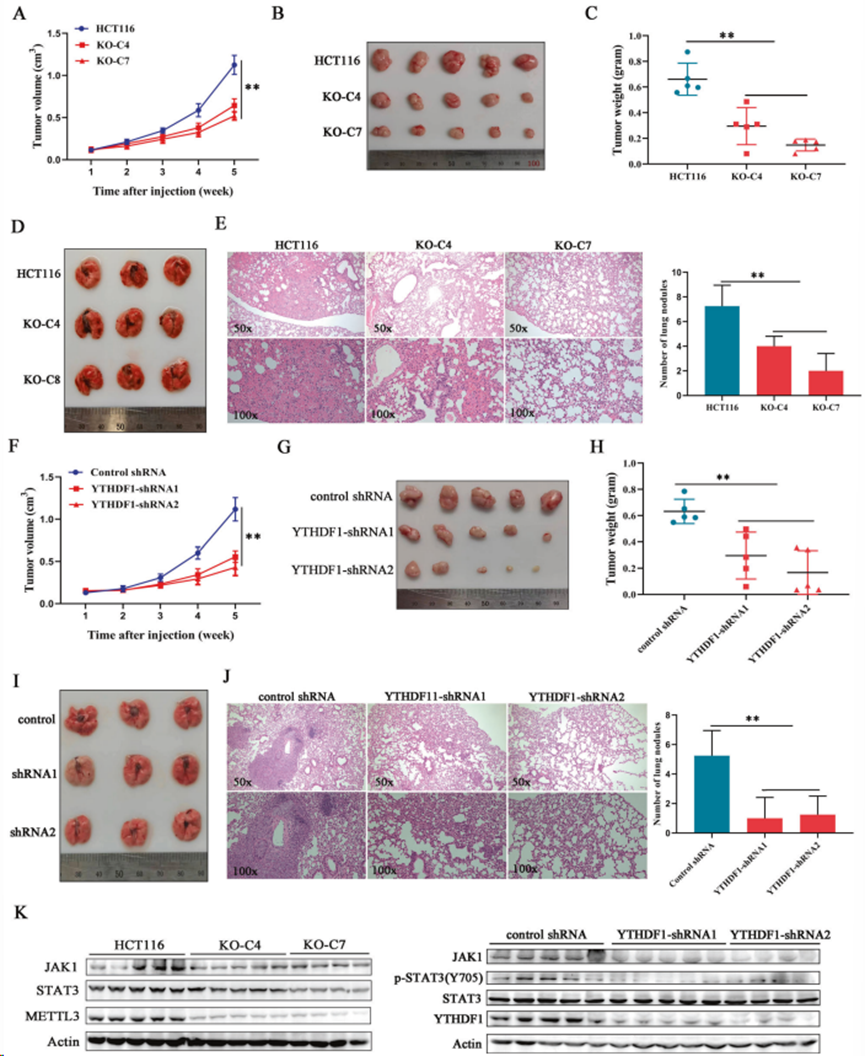
圖7. METTL3和YTHDF1促進體內腫瘤的生長和轉移
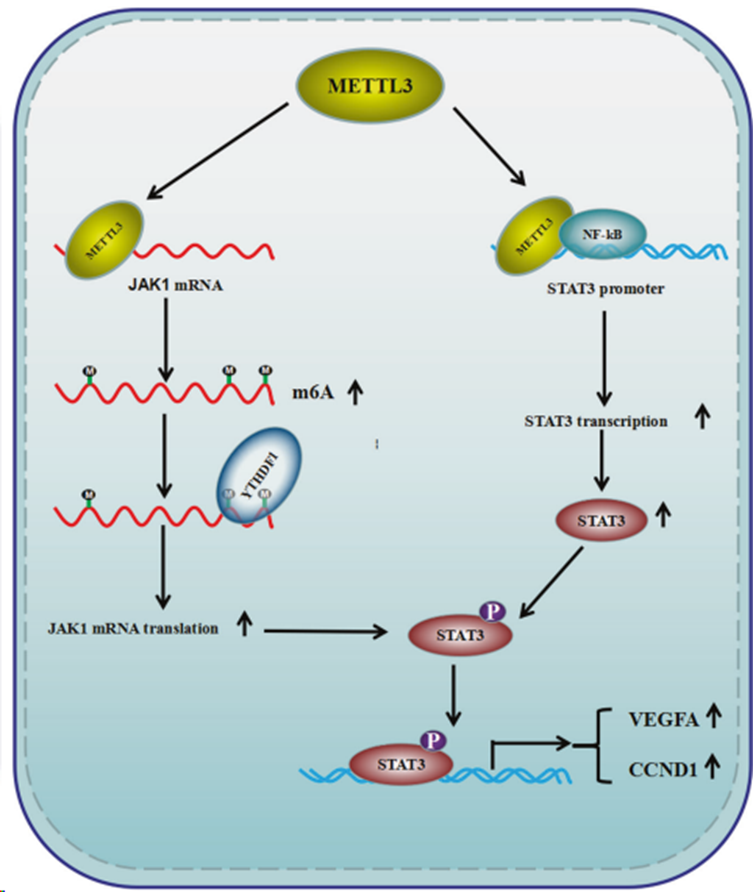
圖8. 結直腸癌細胞中METTL3介導的STAT3信號通路激活示意圖
結論
綜上所述,本研究表明,METTL3通過上調JAK1和STAT3的表達,以m6A 依賴性和非依賴性的方式,促進p-STAT3信號通路的激活,從而促進結直腸癌的進展。因此,同時抑制p-STAT3 和METTL3可為結直腸癌治療帶來新的視角。
實驗方法
細胞培養,細胞增殖和克隆形成實驗,Transwell,細胞周期研究,WB,實時定量PCR,穩定敲低和敲除細胞系的建立,m6A點印跡,m6A RNA免疫沉淀,RNA相關免疫沉淀,ChIP,RNA降解測定,蛋白質穩定性,熒光素酶檢測,動物模型構建
參考文獻
Sun Y, Gong W, Zhang S. METTL3 promotes colorectal cancer progression through activating JAK1/STAT3 signaling pathway. Cell Death Dis. 2023 Nov 25;14(11):765. doi: 10.1038/s41419-023-06287-w. PMID: 38001065; PMCID: PMC10673931.