






ELF5通過穩(wěn)定腎細胞癌中的WDTC1來抑制血管生成
膳食來源的營養(yǎng)素通過提供能量和生物合成構(gòu)件以及作為調(diào)節(jié)分子發(fā)揮作用,與人體生理有著千絲萬縷的聯(lián)系。然而,人體循環(huán)中的營養(yǎng)物質(zhì)影響特定生理過程的機制仍不清楚。在這里,作者使用基于血液營養(yǎng)素化合物庫的篩選方法來證明膳食十八碳烯酸(TVA)在體內(nèi)直接促進效應(yīng)CD8+T細胞功能和抗腫瘤免疫。TVA是母乳中富集的反式脂肪酸的主要形式,但人體不能內(nèi)源性產(chǎn)生TVA。人類循環(huán)中的TVA主要來自反芻動物來源的食物,包括牛肉、羊肉和牛奶、黃油等乳制品,但人類或小鼠分別只有19%或12%的膳食TVA被轉(zhuǎn)化為魯美尼克酸。在機制上,TVA使細胞表面受體GPR43失活,GPR43是一種被其短鏈脂肪酸配體激活的免疫調(diào)節(jié)G蛋白偶聯(lián)受體。因此,TVA拮抗GPR43的短鏈脂肪酸激動劑,導(dǎo)致cAMP-PKA-CREB軸的激活,從而增強CD8+T細胞功能。這些發(fā)現(xiàn)表明,與宿主內(nèi)腸道微生物群來源的短鏈脂肪酸不同,飲食來源的TVA代表了一種宿主-外源性CD8+ T細胞重編程的機制。因此,TVA在腫瘤治療中具有轉(zhuǎn)化潛能。本文于2023年11月發(fā)表于《Nature》,IF: 64.8;Q1。
技術(shù)路線:
主要實驗結(jié)果:
1、增強T細胞功能的營養(yǎng)素
作者利用初始篩選(1a)確定了增強由CD3和CD28抗體(抗CD3/CD28)刺激的Jurkat T細胞活化的營養(yǎng)素(圖1a,頂部)。使用Screen 1b篩選可挽救表達PD - L1的人H596肺癌細胞共培養(yǎng)誘導(dǎo)穩(wěn)定表達PD-1的Jurkat T細胞PD-L1-PD-1依賴性耗竭的營養(yǎng)素(圖1a,底部)。TVA排在首位,并進一步證實其可以增強小鼠和人類原代T細胞的IL-2產(chǎn)生,并挽救共培養(yǎng)表達PD - L1的人類癌細胞誘導(dǎo)的Jurkat T細胞的PD - L1 - PD -1依賴性耗竭。
2、膳食TVA增強抗腫瘤免疫
作者發(fā)現(xiàn)TVA通過T細胞發(fā)揮作用。此外,與喂食對照飲食的小鼠相比,在喂食富含TVA飲食的同系小鼠中,免疫原性B16F10細胞的腫瘤生長潛力顯著減弱(圖1b,頂部)。相比之下,喂食富含CVA的飼料或?qū)φ诊暳系膶φ战MB16F10細胞的腫瘤生長潛力沒有差異(圖1b,底部)。數(shù)據(jù)表明TVA通過調(diào)節(jié)CD8+T細胞來促進抗腫瘤免疫。
3、TVA增強CD8+T細胞功能
流式細胞術(shù)分析顯示,TVA飲食導(dǎo)致腫瘤浸潤淋巴細胞(TILs)中的CD8+T細胞群增加,而對照CVA飲食沒有這一結(jié)果。相比之下,TILs中的CD4+ T細胞群未發(fā)生改變(圖1d)。
TVA飲食導(dǎo)致B16F10腫瘤和dLNs中浸潤的CD45+白細胞中CD8+T細胞的百分比較高,但脾臟中未出現(xiàn)這一情況(圖1e)。耗竭標志物PD-1(圖1f)的表達水平降低,膳食TVA減少了腫瘤和脾臟中CD8+T細胞的耗竭,但在dLNs中無此作用。對具有其他代表性標記的腫瘤浸潤CD8+ T細胞的進一步分析表明,膳食TVA促進CD8+ T細胞功能,增加細胞因子水平,包括TNF(圖1g),增殖標志物Ki-67,共刺激受體ICOS和細胞溶解分子GZMB(圖1h),以及莖樣CD8+T細胞存活標記TCF1(圖1i)。總之,這些結(jié)果表明,膳食TVA促進腫瘤浸潤的CD8+T細胞的聚集和功能。
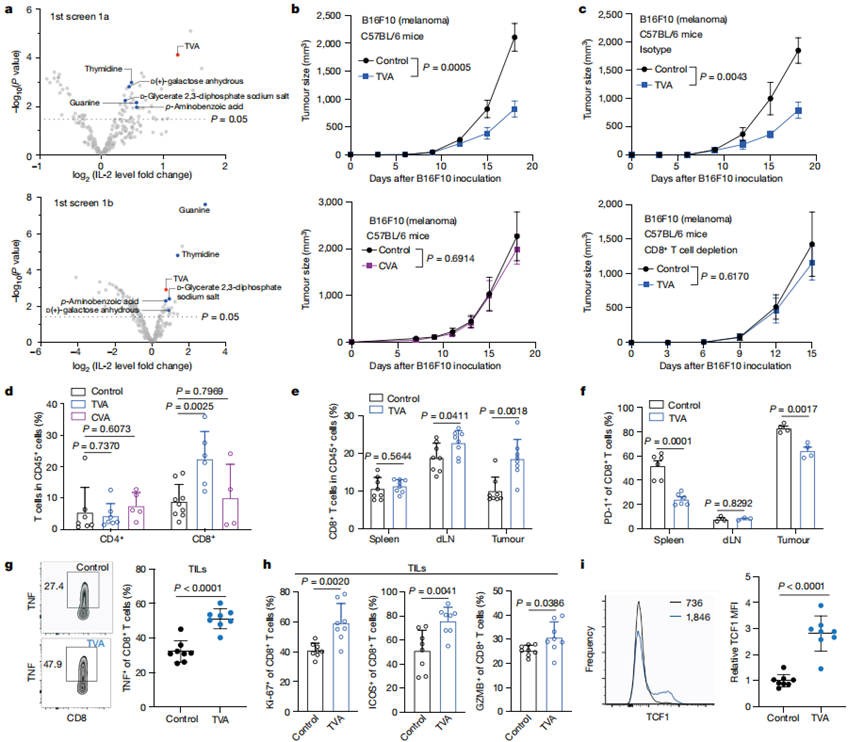
圖1 膳食TVA通過效應(yīng)物CD8+T細胞增強抗腫瘤免疫
4、TVA信號通過GPCR-CREB軸
接下來,作者確定TVA是在細胞外還是細胞內(nèi)發(fā)揮作用。作者使用整合的時間機制研究來研究TVA對人類或小鼠原代CD8+T細胞的影響,包括:(1)酮乙氧基輔助的單鏈DNA測序(KAS-seq)方法,通過捕獲全局轉(zhuǎn)錄動力學(xué)來確定TVA對細胞的初始(20分鐘- 2小時)效應(yīng);(2)磷酸化抗體芯片識別早期(40 min-24 h)細胞信號通路的變化;(3)RNA測序(RNA-seq)方法(24 h),用于全轉(zhuǎn)錄組分析。通過對基因組水平的KAS-seq結(jié)果進行功能富集,發(fā)現(xiàn)TVA處理的CD8+T細胞中富集程度最高的基因體具有GPCR活性(圖2a)。與這一發(fā)現(xiàn)一致的是,早在TVA處理后40分鐘,作者就觀察到跨轉(zhuǎn)錄因子CREB(GPCRs的共同下游效應(yīng)因子)的磷酸化增加(圖2b)。
值得注意的是,TVA處理增強了在效應(yīng)性CD8+T細胞功能中富集的基因E2F(圖2c)的表達,與增強的CD8+T細胞功能和增殖相關(guān)。TVA處理還上調(diào)了Creb1(編碼CREB)以及Prkacb和Prkaca(分別編碼cAMP依賴性蛋白激酶A (PKA)的催化亞基α和β)的表達(圖2d)。綜上所述,這些結(jié)果表明TVA增強CD8+T細胞功能是通過GPCR-CREB途徑介導(dǎo)的,正反饋增強PKA和CREB在基因水平的表達。
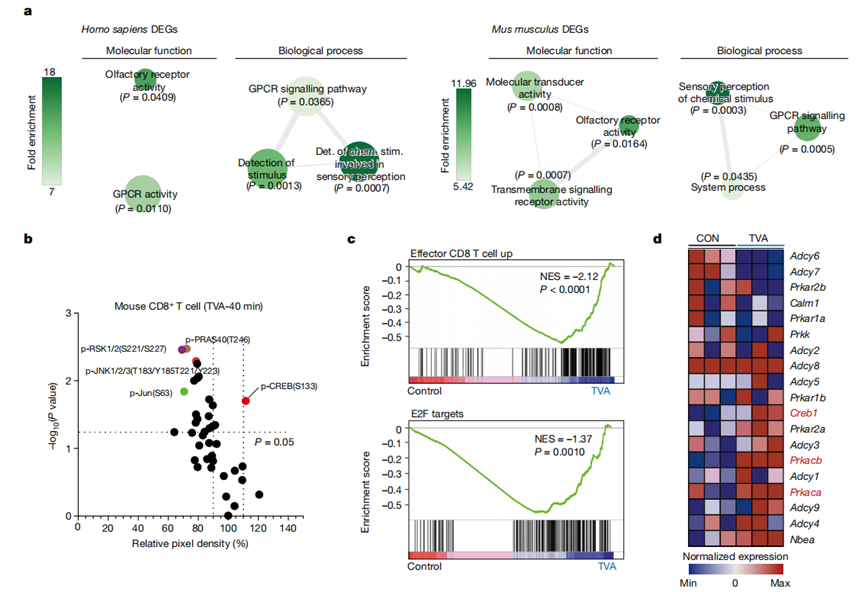
圖2 TVA通過GPCR-CREB軸具有調(diào)節(jié)功能
5、TVA的作用需要CREB
作者使用CD8+T細胞和Creb1敲低對照細胞,在TVA存在或不存在的情況下,用非靶向?qū)φ眨?span>siNTC)短抑制RNA(siRNA)處理。主成分分析表明,在TVA存在和不存在的情況下,使用靶向Creb(siCreb1)的siRNA處理的細胞可被分組在一起,并與siNTC對照細胞組或使用TVA處理的對照細胞組分離(圖3a)。GSEA分析顯示,敲低CREB逆轉(zhuǎn)了與效應(yīng)性CD8+ T細胞功能和細胞增殖相關(guān)的TVA依賴的基因集上調(diào),包括E2F(圖3b)。
為了進一步確定TVA - CREB的下游靶點,作者表征了僅在siNTC + TVA組與其他三個組相比上調(diào)或下調(diào)的基因(圖3c,左側(cè))。共312個上調(diào)基因富集在11個GO類別,包括細胞周期、細胞增殖、細胞分裂、T細胞聚集、T細胞活化、T細胞差異分化和細胞因子產(chǎn)生(圖3c,右上)。相比之下,共有139個下調(diào)基因被富集在8個GO類別中,包括凋亡和染色質(zhì)組織(圖3c,右下)。為了進行功能驗證,作者選擇了4個在細胞增殖和T細胞功能中至關(guān)重要的上調(diào)基因。這些基因包括Il18(與細胞增殖、T細胞活化和細胞因子產(chǎn)生相關(guān))、Ebi3(與細胞增殖和T細胞活化相關(guān))、Tbx21(與T細胞活化和細胞因子產(chǎn)生相關(guān))和Ilf2(與細胞增殖、T細胞活化和細胞因子產(chǎn)生相關(guān))。作者還測試了Foxo4和Bcl6,它們是與凋亡相關(guān)的GO分類中的關(guān)鍵基因。
如圖3d所示,與siNTC處理的對照細胞相比,通過siCreb1敲低Creb1的表達有效地逆轉(zhuǎn)了TVA依賴的細胞數(shù)量和凋亡,以及CD8+T細胞中Ki-67、IL-2、TNF和IFNγ水平的變化。敲低Il18、Ebi3、Tbx21、Ilf2、Foxo4或Bcl6部分逆轉(zhuǎn)了TVA依賴性細胞數(shù)量的變化(圖3d,左)。Bcl6、Foxo4或Ebi3的敲低部分逆轉(zhuǎn)了CD8+T細胞凋亡群中的TVA依賴性變化,而Il18、Tbx21或Ilf2的敲低沒有這種作用(圖3d,中)。敲低Il18、Tbx21、Ilf2或Ebi3部分逆轉(zhuǎn)了Ki-67水平的TVA依賴性變化,而敲低Bcl6或Foxo4沒有這種作用(圖3d,右)。總之,這些結(jié)果確立了不同的CREB靶基因?qū)?span>CD8+T細胞中不同的TVA依賴變化的功能貢獻。
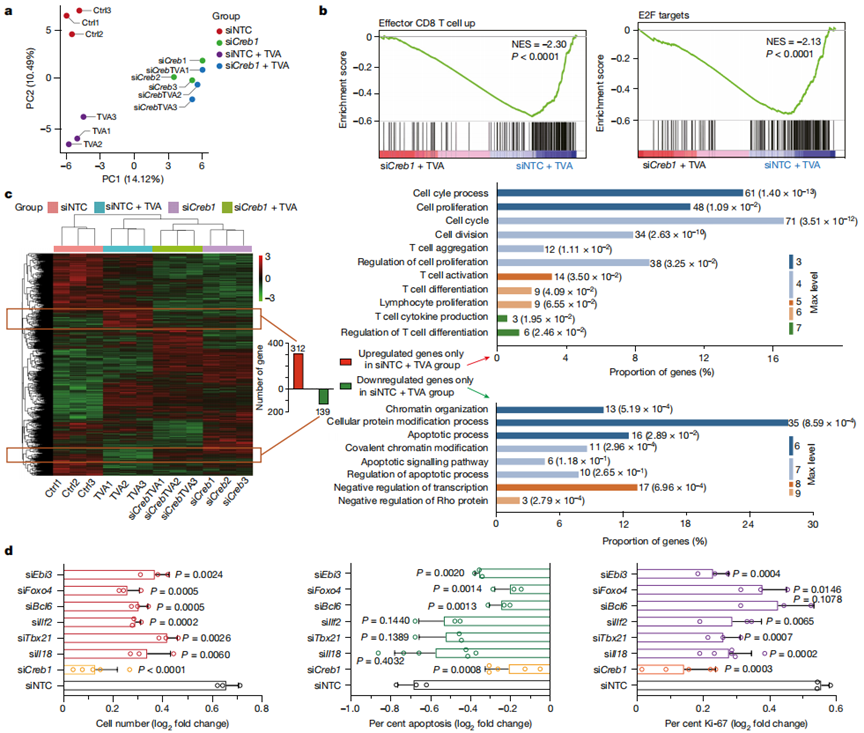
圖3 TVA對CD8+ T細胞的作用主要通過CREB及其靶基因集介導(dǎo)
6、TVA使結(jié)合SCFA的GPR43失活
為了確定TVA的GPCR靶點,作者通過siRNA介導(dǎo)的敲低篩選了6個已知的脂肪酸結(jié)合GPCRs,其中,只有敲低GPR43才能消除原代小鼠CD8+T細胞中TVA增強的TNF水平(圖4a)。使用siRNA介導(dǎo)的GPR43敲低的OT-I T細胞和使用CRISPR-Cas9敲低GPR43的OT-I T細胞(圖4b)獲得了類似結(jié)果。這些數(shù)據(jù)表明,GPR43在CD8+T細胞活化中具有抑制作用,TVA可能減弱GPR43的功能。這可能部分解釋了活化的CD8+T細胞對TVA治療敏感的原因。
接下來,作者對TVA進行了構(gòu)效關(guān)系(SAR)研究。SAR研究的結(jié)果表明,雙鍵(1),酸基(2)和長度至少16個碳(3,4,13,15和16)是維持TVA生物活性的關(guān)鍵。此外,將雙鍵的位置從C11-C12轉(zhuǎn)移到C9-C10(6),或添加一個C9-C10雙鍵(7),都會增強TVA的生物活性,表明C9-C10雙鍵可能是最適合TVA生物活性的雙鍵。對9、10、14和17的SAR結(jié)果也表明,末端鏈可以進行修飾,得到的TVA衍生物保留了70-80%的生物活性。接下來,作者使用經(jīng)TVA探針3處理的小鼠CD8+T細胞進行了光親和標記研究。重氮嘧啶使之能夠共價光交聯(lián)到接觸蛋白殘基并且炔基使之能夠連接報告子(生物素)并探測TVA結(jié)合蛋白靶點。使用鏈親和素珠下拉的生物素標記蛋白的蛋白質(zhì)印跡顯示TVA探針與GPR43之間的結(jié)合(圖4c)。
作者接著發(fā)現(xiàn),通過降低TNF(圖4d)水平評估,將醋酸SCFA濃度增加至20 mM可減弱CD8+T細胞活化。20 μM TVA有效地逆轉(zhuǎn)了這一讀數(shù)。相比之下,2 μM TVA足以逆轉(zhuǎn)20 mM醋酸鹽對CD8+T細胞的抑制(圖4e)。相反,20 μM TVA預(yù)處理增強的CD8+ T細胞活化不能被20 mM醋酸鹽逆轉(zhuǎn)。這些結(jié)果表明,TVA可能通過拮抗GPR43的SCFA激動劑結(jié)合并失活GPR43。
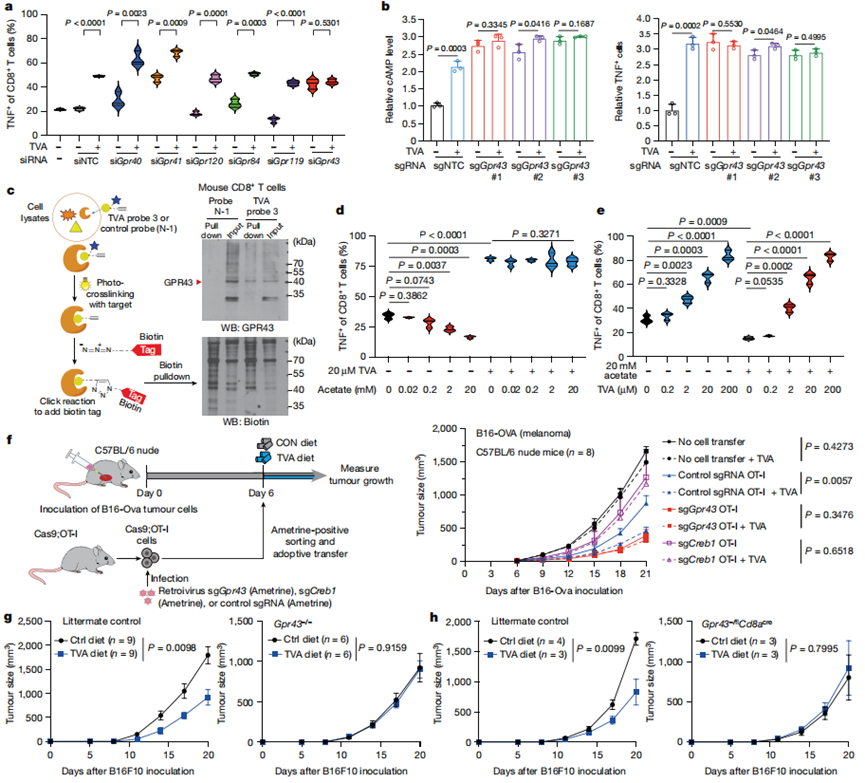
圖4 TVA使SCFA結(jié)合的GPR43失活
7、TVA活性的GPR43要求
作者首先使用表達cas9的小鼠OT-I T細胞檢測過繼性細胞治療(ACT)小鼠模型。作者使用接種了表達同源抗原(B16-OVA)的小鼠B16F10黑色素瘤細胞的同系小鼠作為OT-I T細胞的受體,以確定抗腫瘤免疫(圖4f,左)。使用對照單向?qū)?span>RNA(sgRNA)過繼轉(zhuǎn)移OT-I細胞導(dǎo)致腫瘤生長降低,使用富含TVA的飲食進一步降低了腫瘤生長(圖4f,右)。相比之下,使用CRISPR - Cas9介導(dǎo)的Gpr43敲除過繼轉(zhuǎn)移OT-I細胞顯著降低了腫瘤生長,而TVA飲食無法進一步抑制腫瘤生長(圖4f,右)。與這些發(fā)現(xiàn)一致,與對照OT-I細胞相比,采用CRISPR - Cas9介導(dǎo)的Creb1敲除的OT-I細胞過繼轉(zhuǎn)移導(dǎo)致小鼠的抗腫瘤效應(yīng)減弱,并且對TVA飲食無應(yīng)答(圖4f,右)。
接下來,作者使用接種小鼠B16F10細胞的全身Gpr43敲除(Gpr43 - / -)小鼠進行同系小鼠模型實驗。作者發(fā)現(xiàn)Gpr43 - / -小鼠的GPR43缺乏(通過Gpr43 mRNA水平在CD8+和CD4+T細胞以及B細胞中得到證實)消除了在同窩對照小鼠中觀察到的TVA飲食依賴性腫瘤生長減少(圖4g)。
CD8+T細胞中Gpr43的條件性敲除消除了在同窩對照小鼠中觀察到的TVA飲食依賴性腫瘤生長的減少。綜上所述,這些結(jié)果表明TVA需要CD8+T細胞中的GPR43來增強CD8+T細胞的功能,從而在體內(nèi)產(chǎn)生抗腫瘤免疫。
8、TVA拮抗SCFAs對cAMP的作用
培養(yǎng)液中的SCFAs激活CD8+ T細胞中的GPR43,并使其易于發(fā)生TVA介導(dǎo)的失活。因此,作者比較了TVA和SCFAs對CD8+T細胞cAMP信號傳導(dǎo)的相反作用。作者首先研究了SCFAs是否通過GPR41和GPR43以及pH感應(yīng)器GPR65抑制CD8+ T細胞活性,因為與TVA等長鏈脂肪酸不同,SCFAs可溶于水并降低管腔pH。GPR65可被低pH激活,并通過Gαs發(fā)出信號,增加cAMP的產(chǎn)生以及隨后CREB的磷酸化和激活。CD8+ T細胞活性的增加可被SCFA部分抑制。相比之下,敲低GPR65導(dǎo)致CD8+T細胞活性下降,而SCFA進一步降低,而敲低GPR41, GPR43和GPR65則完全消除了SCFA對CD8+T細胞的作用。相比之下,TVA在兩種檢測中均顯示出最小的影響。總之,SCFAs抑制總體CD8+ T細胞活性,即GPR41和GPR43介導(dǎo)的cAMP負性調(diào)節(jié)可拮抗GPR65對cAMP水平的積極作用。
相比之下,作者發(fā)現(xiàn)TVA飲食不會改變來自對照或TVA飲食喂養(yǎng)的小鼠的血清和TIF樣本中的pH,并且在來自具有不同TVA血清水平的一組淋巴瘤患者的人類初級血清樣本中沒有檢測到pH的顯著差異(詳細描述見圖5d)。最后,在使用表達Cas9的OT-I T細胞的ACT小鼠模型中,CRISPR - Cas9介導(dǎo)的Gpr65敲除導(dǎo)致腫瘤生長下降,然而,TVA飲食進一步降低了腫瘤生長(注意到前4個對照組來自圖4f中描述的相同實驗)。這些結(jié)果表明,在體外和體內(nèi),TVA不會降低pH值,也不會通過GPR65發(fā)出信號來增強CD8+T細胞功能,這支持作者的假設(shè),即TVA使GPR43失活,并拮抗SCFAs對cAMP的總體負面影響。
9、TVA可增強基于T細胞的療法
膳食TVA聯(lián)合抗PD -1抗體(免疫檢查點抑制劑治療的代表)顯示出對B16F10腫瘤生長的協(xié)同抑制作用(圖5a)。作者接下來測試了TVA對博納吐單抗(blinatumomab)療效的影響,博納吐單抗是一種靶向B細胞上的CD19和T細胞上的CD3的雙特異性T細胞銜接器。在博納吐單抗存在的情況下,TVA以劑量依賴性方式顯著增強了人外周血單個核細胞(PBMCs)對人B-ALL RS4;11細胞的體外殺傷效率(圖5b)。此外,TVA增加了來自3例42 ~ 47歲淋巴瘤患者原代T細胞的嵌合抗原受體(CAR)T細胞的體外擴增(圖5c)。值得注意的是,在一項回顧性臨床研究中,作者發(fā)現(xiàn),對CAR-T細胞療法有應(yīng)答的淋巴瘤淋巴瘤患者組的血清TVA水平高于對CAR-T細胞療法無應(yīng)答的患者組(圖5d)。這些發(fā)現(xiàn)與以下觀點一致:通過膳食攝入TVA可能提高對T細胞免疫療法的臨床應(yīng)答。
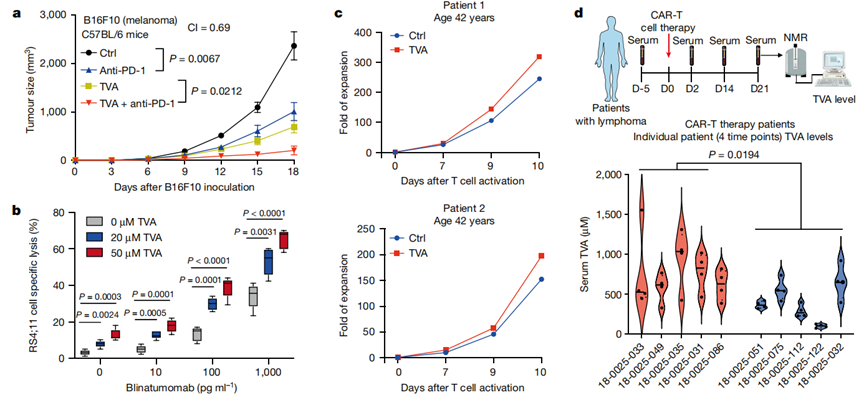
圖5 TVA增強了多種基于T細胞的抗癌療法的有效性
結(jié)論:
作者的研究結(jié)果揭示了人類飲食進化過程中的一種機制,即生物體外的TVA通過外源性調(diào)節(jié)重新編程CD8+T細胞,從而使GPR43失活,這與作為GPR43激動劑的腸道微生物群來源的生物體內(nèi)SCFAs不同。TVA具有很高的轉(zhuǎn)化潛力,可以作為一種膳食元素,改善多種抗腫瘤療法的臨床療效,如免疫檢查點抑制劑,T細胞銜接器,CAR-T和T細胞受體T細胞療法。作者的研究支持補充TVA是一種比改變飲食更有針對性和有效的方式,有利于抗腫瘤免疫。GPR43-CREB機制可能對CD8+T細胞具有細胞類型特異性。最后,由于TVA與SCFAs相比體積較大,可能結(jié)合到一個不同的位點并作為負變構(gòu)調(diào)節(jié)劑發(fā)揮作用。需要進一步的研究來闡明TVA使GPR43失活的潛在結(jié)構(gòu)和分子機制。
實驗方法:
PD-1+ Jurkat T細胞系構(gòu)建;循環(huán)營養(yǎng)庫篩選;小鼠腫瘤模型;飲食方案;抗體介導(dǎo)的T細胞耗竭;分泌細胞因子水平;Pmel-1殺傷實驗;細胞增殖實驗;核磁共振法提取和定量TVA水平;CD45+腫瘤浸潤性白細胞分離;小鼠TIL分離;小鼠脾淋巴細胞分離;小鼠dLNs淋巴細胞分離;主要CD8+或CD4+T細胞分離和激活;流式細胞術(shù);抗體;微生物組16S測序;[13C1]氣相色譜-質(zhì)譜法分析TVA代謝通量;細胞培養(yǎng)處理;KAS-seq和數(shù)據(jù)分析;實時定量PCR;RNA介導(dǎo)的細胞siRNA干擾;RNA測序;[13C]脂肪酸的體外示蹤;海馬脂肪酸氧化試驗;小鼠OT-I細胞的CRISPR編輯;Pull-down試驗鑒定交聯(lián)蛋白-TVA復(fù)合物;與Blinatumomab共培養(yǎng)試驗;CAR-T細胞擴增試驗。
參考文獻:
Fan H, Xia S, Xiang J, Li Y, Ross MO, Lim SA, Yang F, Tu J, Xie L, Dougherty U, Zhang FQ, Zheng Z, Zhang R, Wu R, Dong L, Su R, Chen X, Althaus T, Riedell PA, Jonker PB, Muir A, Lesinski GB, Rafiq S, Dhodapkar MV, Stock W, Odenike O, Patel AA, Opferman J, Tsuji T, Matsuzaki J, Shah H, Faubert B, Elf SE, Layden B, Bissonnette BM, He YY, Kline J, Mao H, Odunsi K, Gao X, Chi H, He C, Chen J. Trans-vaccenic acid reprograms CD8+ T cells and anti-tumour immunity. Nature. 2023 Nov 22. doi: 10.1038/s41586-023-06749-3. Epub ahead of print. PMID: 37993715.