






IGF2BP3通過m6A RNA甲基化調節喉癌中TMA7介導的自噬和順鉑耐藥
翻譯體系相關的7同源物(TMA7)與增殖相關疾病密切相關。然而,TMA7在喉鱗狀細胞癌(LSCC)中的作用和調控機制尚不清楚。本研究旨在探討TMA7在喉癌發生發展中的作用,并探討其作用機制。TMA7在喉鱗狀細胞癌組織中表達上調,并與預后不良有關。TMA7下調后,自噬水平增加,抑制了LSCC細胞的增殖、遷移和侵襲。M6A甲基化閱讀器IGF2BP3增強了TMA7的穩定性,降低了自噬水平。TMA7與UBA2直接相互作用。此外,IGF2BP3調節的TMA7-UBA2-PI3K通路的激活是TMA7抑制自噬、促進喉癌進展的主要機制。目前的研究表明,IGF2BP3介導的TMA7 m6A修飾通過UBA2-PI3K途徑促進LSCC的進展和順鉑耐藥,為LSCC自噬相關機制、潛在的生物標志物和治療靶點提供了新的見解。本文于2023年2月發表于《International Journal of Biological Sciences》期刊上,IF=9.2
主要技術路線:
1、喉癌組織中TMA7癌基因表達上調
為了研究該基因的表達,我們選擇了5對喉癌和癌旁組織進行蛋白質組學和轉錄組學聯合分析。我們選擇了OmicStudio工具來分析數據。結果如圖1a-e所示。在log2FoldChange>1或log2FoldChange<-1(p<0.05)的轉錄分析中,剔除極端數據后,選取有代表性的基因進行聚類分析,其中54個基因與癌旁組織相比顯著上調,11個基因顯著下調(圖1A)。在蛋白質組學分析結果中,在p<0.05處,2486個蛋白質表現出顯著差異。在相同條件下,剔除極端數據后,選擇具有代表性的蛋白質進行聚類分析,與癌旁組織相比,15個蛋白質顯著上調,15個蛋白質顯著下調(圖1B)。
轉錄組分析表明,6828個基因在患者樣本中與鄰近組織中存在差異表達。在蛋白質組學數據中,共有6187個蛋白質得到了差異表達。在兩組數據中總共有2131個差異表達的基因(圖1C)。對這兩組數據的相關性分析表明,在轉錄和蛋白質組數據中存在差異表達的基因,如圖1D所示。藍點代表了兩個具有相同組學表達趨勢的基因。火山圖也是使用OmicStudio工具進行的(圖1E)。此外,我們評估了在特定篩選條件下喉癌組織中上調基因的意義、組織中的蛋白質含量以及差異表達倍數,并鑒定了上調表達的基因。
其中,TMA7在RNA和蛋白質水平上均有上調。因此,我們選擇TMA7作為目的基因。統計分析表明,喉癌組織中TMA7的表達水平在蛋白質水平和RNA水平均高于癌旁組織。根據喉癌組織中TMA7的表達情況分析其臨床病理特征(表1)。如表1所示,喉癌TMA7高表達患者的臨床分期(***p<0.001)和T分期(**p<0.01)均較高。此外,TMA7升高的患者有淋巴結轉移(***p<0.001)和頻繁復發(**p<0.01)。然而,TMA7的表達與年齡、性別和原發部位無關。此外,Kaplan-Meier生存曲線顯示TMA7升高與預后不良相關(圖1F-G)。因此,TMA7的IHC是在喉癌和癌旁組織中進行的,如圖1H所示。
圖1 TMA7作為癌基因在喉癌組織中表達上調
2、TMA7影響喉鱗狀細胞癌的增殖、侵襲、遷移和凋亡
為了闡明TMA7在LSCC細胞中的生物學功能,我們用慢病毒TU177、AMC - HN8和TU212的shRNA轉染LSCC細胞系來KD TMA7的表達(TMA7-KD)。RT-qPCR和Western blot證實TMA7被成功敲除(圖2A, B)。集落形成和EdU檢測顯示TMA7-KD抑制細胞增殖(圖2C, D)。Transwell分析顯示,TMA7 KD降低了LSCC細胞的侵襲性(圖2e)。隨后,通過傷口愈合試驗進行評估了LSCC細胞的遷移能力降低,隨后TMA7水平下降(圖2F)。在LSCC細胞TMA7-KD后,細胞的凋亡率增加(圖2G)。綜上所述,這些結果表明TMA7-KD減弱了LSCC細胞的增殖、侵襲、遷移和集落形成能力,增加了細胞的凋亡率。
圖2 TMA7影響喉鱗狀細胞癌的增殖、侵襲、遷移和凋亡
3、TMA7與喉癌細胞自噬相關
為了研究TMA7與下游分子之間的調控相關性,我們對三個LSCC細胞系及其相應的TMA7 KD細胞系進行了RNA-SEQ檢測。相關下游RNA分子的表達水平發生了變化。我們還發現,在FC≥1.5且p<0.05的截止點TMA7-KD上調了945個基因,下調了2521個基因。接下來,我們篩選出了泛素樣修飾物激活酶2(UBA2),它通過添加小蛋白SUMO(參見SUMO 1;MIM 601912)或類泛素化來調節蛋白質結構和細胞內定位,從而實現蛋白質的翻譯后修飾。我們發現它的mRNA水平降低,經過GO分析,它與蛋白質結合有關。數據庫分析表明,它在UniProt預測的細胞質中表達。UBA2-KD促進腎透明細胞癌細胞的凋亡。本研究討論了與細胞凋亡和自噬相關的表型,因為細胞凋亡和自噬與癌癥有關。因此,我們推測UBA2除了與細胞凋亡有關外,是否還與自噬有關,這在以前還沒有描述過。本研究探討了UBA2與自噬表型相關途徑的調控關系,并選擇UBA2作為TMA7的下游基因進行進一步研究。
為了研究喉鱗狀細胞癌TMA7-OE細胞生物學功能的變化,將TMA7-OE慢病毒導入LSCC細胞。RT-qPCR結果顯示,TMA7-OE慢病毒感染LSCC細胞后,TMA7的表達相應增加(圖3A)。TMA7-OE慢病毒從Genecem(MD,美國)獲得。此外,我們進行了Western印跡以驗證LSCC細胞TMA7的OE和KD(圖3B)。結果表明,TMA7在LSCC細胞中成功地過表達或沉默(圖2B,圖8A),蛋白質含量相應地增加和減少。為了研究LSCC細胞中的自噬和TMA7,我們檢測了與自噬相關的蛋白:P62、LC3B-I和LC3B-II。TMA7-OE后,細胞自噬減弱,p62表達增加,LC3B-II/I比值降低。TMA7-KD提供了相互矛盾的結果(圖3B)。
用黃色自噬小體(mCherry和GFP)和紅色自噬溶體(MCherry)標記的串聯熒光報告基因(mCherryGFP-LC3)直接監測自噬通量。因此,黃點和紅點分別表示自噬小體和自噬溶體。用腺病毒(mCherry-GFP-LC3、韓恒、中國)感染用TMA7OE或KD感染的LSCC和對照細胞。共聚焦顯微鏡顯示,單個細胞的黃點和紅點的比例不變,但點數增加。分析表明,自噬-溶酶體代謝沒有改變,總體自噬水平增加。TMA7-KD后自噬增加,TMA7-OE后自噬減少(圖3C)。UBA2的表達在TMA7-KD后減少,TMA7-OE后增加(圖3B)。因此,我們推測UBA2可能是TMA7的下游分子,并被選中進行進一步的研究。
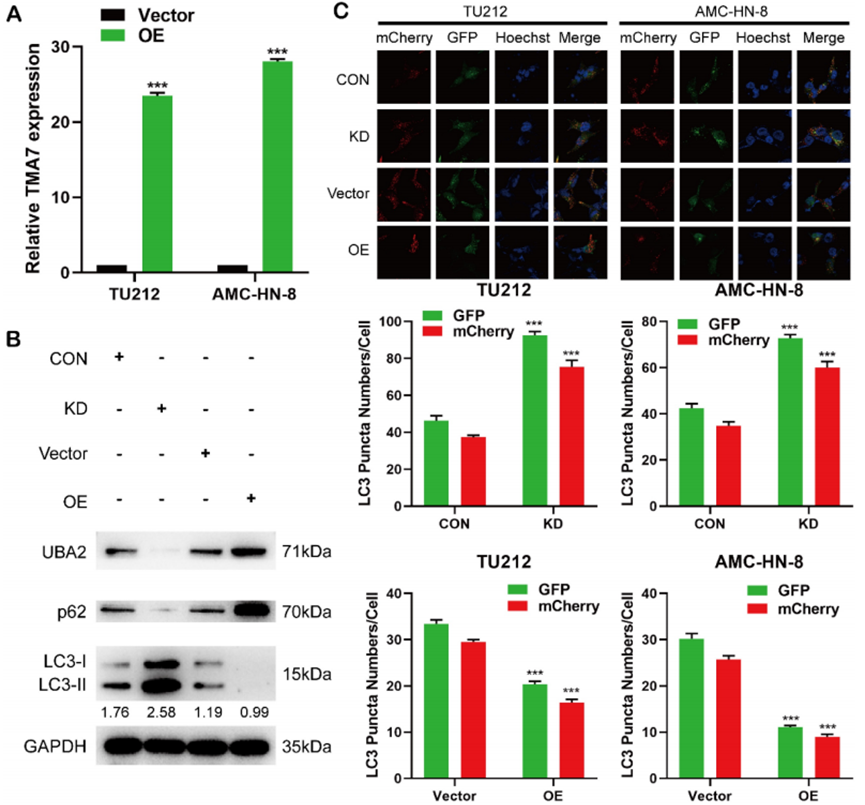
圖3 TMA7與喉鱗狀細胞癌自噬相關
4、IGF2BP3在喉癌細胞中的癌基因功能
對來自多個數據集的差異表達基因的分析表明,除了TMA7上調外,IGF2BP3在喉癌組織中也上調。在LSCC細胞中,IGF2BP3-KD或-OE后,IGF2BP3水平成功改變(圖4A-B)。免疫印跡顯示IGF2BP3-KD后IGF2BP3、TMA7和UBA2表達降低,而IGF2BP3-OE后IGF2BP3、TMA7和UBA2表達增加。為了研究IGF2BP3與LSCC細胞自噬的相關性,我們檢測了自噬相關蛋白P62和LC3B。IGF2BP3-OE后,自噬減弱,p62增加,Lc3B-II/I比值降低。相反,IGF2BP3的下調導致了相反的結果(圖4C)。
接下來,我們研究了IGF2BP3對LSCC細胞增殖、遷移、侵襲和凋亡的影響。克隆形成、EDU、傷口愈合和Transwell分析表明IGF2BP3-KD抑制LSCC細胞的增殖、遷移和侵襲(圖4D-f,4h),而IGF2BP3-OE促進相反的結果。IGF2BP3-KD后LSCC細胞的凋亡增加,IGF2BP3-OE后LSCC細胞的凋亡減少(圖4G)。IGF2BP3-KD后自噬水平增強,IGF2BP3-OE后自噬水平減弱(圖4I)。
IGF2BP3促進LSCC細胞遷移、增殖和侵襲,減少細胞凋亡和自噬,調節TMA7和UBA2的表達水平。
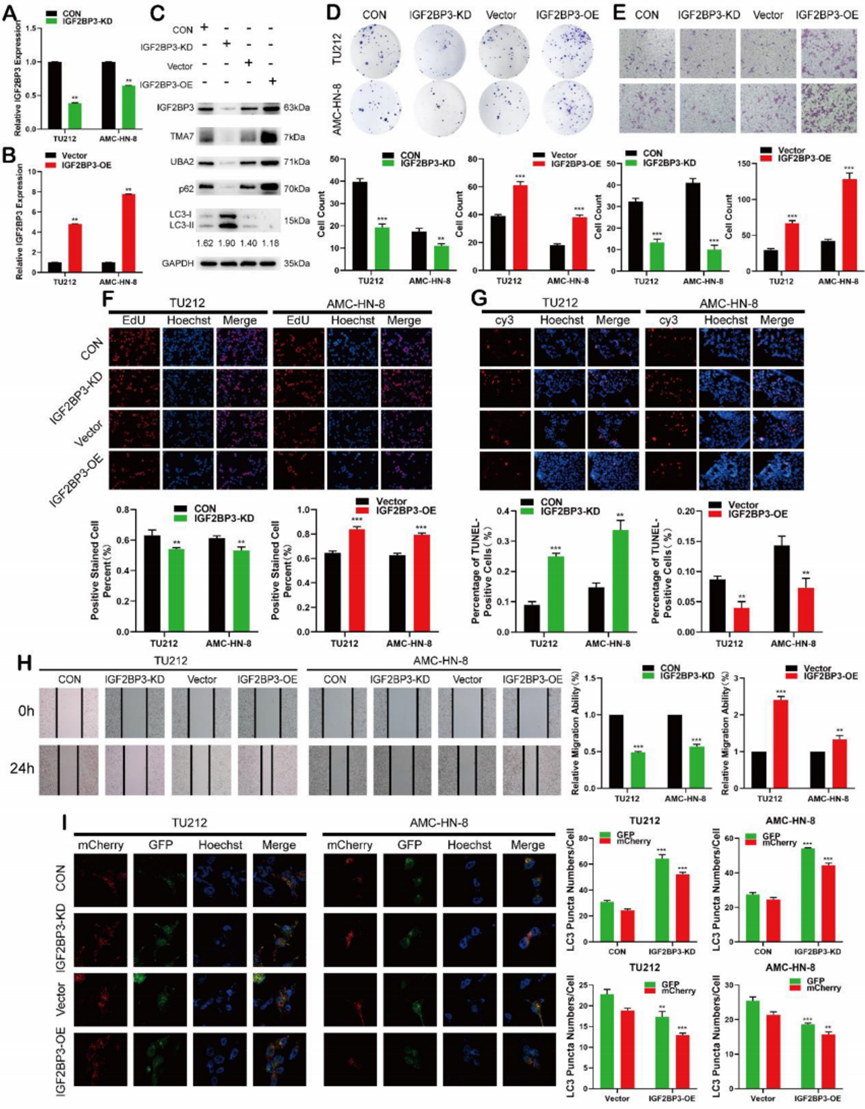
圖4 IGF2BP3在喉癌細胞中起癌基因的作用
5、IGF2BP3通過m6A甲基化調控TMA7
m6A在mRNA調控中起重要作用。TMA7 mRNA的3’-未翻譯(UTR)序列包含一個典型的m6A修飾基序,該基序由生物信息學分析(圖5A)管理。為了預測m6A修飾的可能位點,我們對MEME進行了基序預測,得到了預測的m6A修飾位點序列RRACH(D=A、G或U;R=A或G;H=A、U或C)。m6A甲基化修飾發生在基序的A堿基上(圖5B)。Western印跡分析表明,在TU212細胞中的TMA7正義RNA探針下拉樣本中存在IGF2BP3(圖5C)。為了證明IGF2BP3與TMA7之間的相互作用,我們進行了RIP實驗,結果表明IGF2BP3可能與TMA7結合(圖5D)。接下來,我們在TU212中進行了MERIP,并在TMA7的3’-UTR區域發現了一個m6A修飾位點。因此,我們選擇了上述序列并進行了下一步的驗證。MERIP后的RT-qPCR分析(m6A RIP)進一步證實了m6A修飾的TMA7 mRNA(圖5E)。與促進蛋白質翻譯的潛在作用相一致,減少TMA7 mRNA的m6A修飾降低了TMA7蛋白的表達和新生TMA7蛋白的合成。(圖4C)。隨后,我們得出結論:IGF2BP3直接與TMA7的mRNA分子結合。IGF2BP3 OE或KD后,用Actinomycin D檢測TMA7 mRNA在LSCC細胞上的穩定性。如前所述,提取總RNA用于RT-qPCR分析。計算指定時間點的mRNA表達,并根據GAPDH的表達進行標準化。因此,我們得出結論,IGF2BP3的消除可以縮短TMA7 mRNA的半衰期。IGF2BP3-OE延長了TMA7 mRNA的半衰期(圖5F,g)。
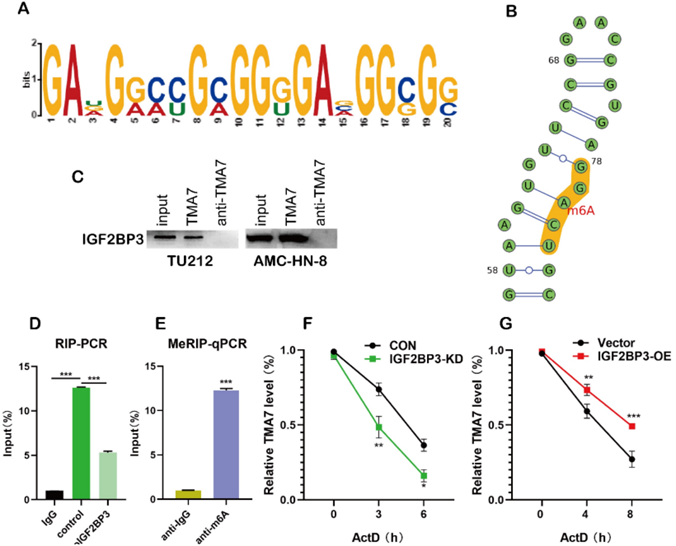
圖5 IGF2BP3通過m6A甲基化識別和結合TMA7
6、UBA2在喉癌細胞中的癌基因功能
在用RNA-seq(圖3)分析了差異表達基因后,我們選擇UBA2作為TMA7的下游分子,用于TMA7-KD之后的下一步研究。
據報道,UBA2在癌癥中起主要作用,但它在喉癌中的作用尚不清楚。目前的數據表明,LSCC細胞中的UBA2-OE或UBA2-KD成功地改變了UBA2的表達(圖6A-C)。為了闡明UBA2和自噬在LSCC細胞中的作用,我們對自噬相關蛋白P62和LC3B進行了研究。UBA2的OE減少了自噬,增加了p62,降低了LC3B-II/I比值。UBA2的破壞導致LC3B-II/I升高,p62降低(圖6C)。
接下來,我們研究了UBA2對LSCC細胞增殖、遷移、侵襲和凋亡的影響。克隆形成、EDU、傷口愈合和Transwell分析表明,UBA2-KD抑制LSCC細胞的增殖、遷移和侵襲,而UBA2-OE促進這種差異(圖6D-G)。UBA2-KD后細胞凋亡率增加,UBA2-OE后細胞凋亡率降低(圖6H)。此外,當UBA2缺失時,自噬增加,而當UBA2過度表達時,自噬減弱(圖6I)。先前的一項研究證明,TMA7的水平與UBA2(圖3B)有關;因此,我們進行了co-IP實驗。結果表明,這兩個分子直接相互作用(圖6J),表明兩個分子可能相互結合。結果表明,UBA2在喉鱗狀細胞癌中起癌基因作用。在UBA2和TMA7之間建立了結合關系。
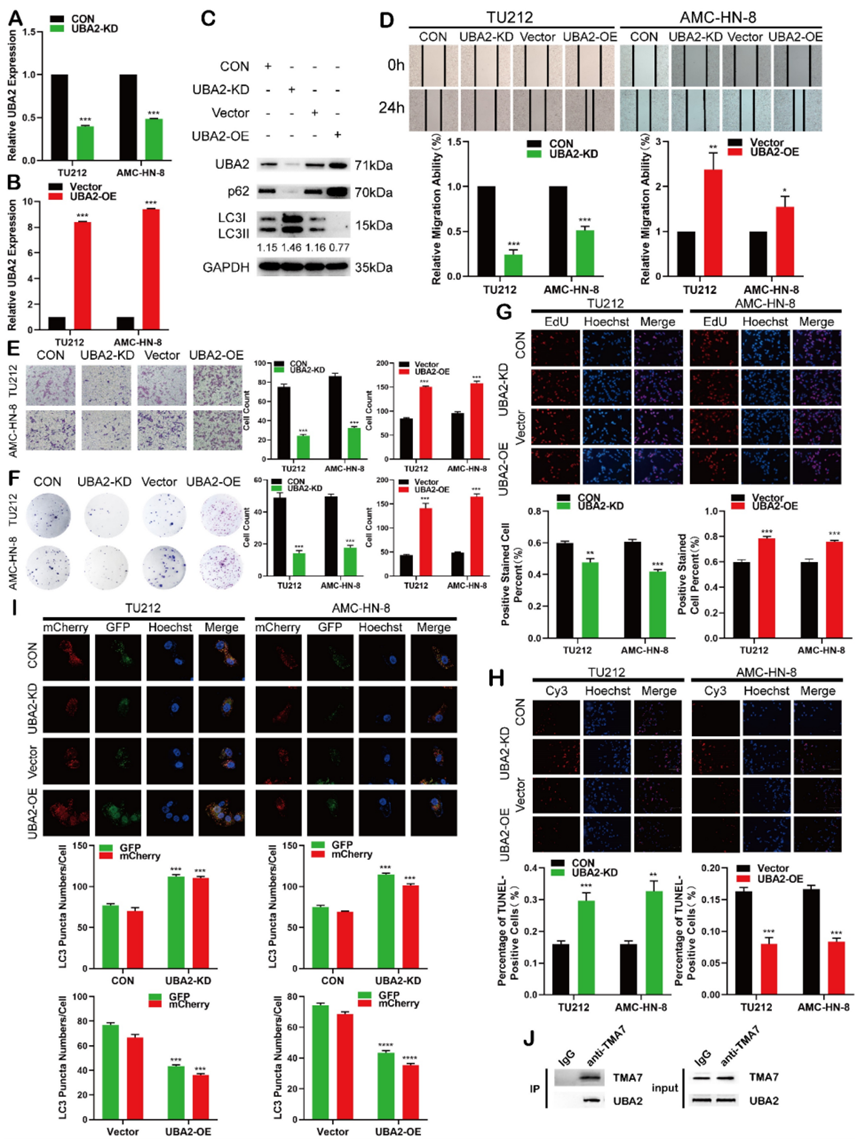
圖6 UBA2在喉癌細胞中的癌基因功能
7、TMA7通過UBA2和PI3K/mTOR途徑影響LSCC自噬
在TMA7-KD后,我們用RNA-seq富集PI3K-mTOR通路,發現PI3K-mTOR通路與TMA7的變化有關。因此,我們進一步研究了TMA7對PI3K-mTOR信號通路的影響。Western印跡分析表明,TMA7-KD減少,而TMA7-OE增加了PI3K和mTOR的磷酸化水平(圖7A)。推測TMA7可以激活PI3K-mTOR信號通路。自噬通量分析表明,UBA2-OE減弱了TMA7-KD引起的自噬增加,而UBA2-KD增強了TMA7-OE引起的自噬減少(圖7C)。
為了評估PI3K-mTOR信號通路在TMA7介導的自噬抑制中的重要性,我們用雷帕霉素處理TMA7-OE LSCC細胞。Western印跡和自噬通量分析表明,當3-MA處理TMA7-KD LSCC細胞時,抑制PI3K磷酸化可以重新激活被TMA7-OE抑制的自噬,而PI3K磷酸化的激活再一次抑制了由TMA7-KD激活的自噬(圖7B,C)。此外,UBA2-OE還可挽救TMA7-KD引起的細胞增殖、侵襲和遷移的減少。此外,UBA2-KD挽救了TMA7-OE引起的細胞增殖、侵襲和遷移(圖7D,7F-H)。UBA2-OE挽救了TMA7-KD引起的細胞凋亡增加,而UBA2-KD拯救了TMA7-OE引起的細胞凋亡減少(圖7E)。因此,我們推測TMA7與UBA2相互作用,并通過PI3K/mTOR信號通路調節喉癌自噬水平。

圖7 TMA7通過UBA2和PI3K/mTOR途徑影響LSCC自噬
8、IGF2BP3通過TMA7調節LSCC自噬和UBA2表達
為了驗證TMA7在IGF2BP3介導的抑制自噬中的重要性,用TMA7-KD感染IGF2BP3-OE LSCC細胞,用TMA7-OE慢病毒處理IGF2BP3-KD LSCC細胞。Western印跡顯示,TMA7-KD可抑制IGF2BP3-OE誘導的UBA2上調和自噬抑制。而IGF2BP3-KD對UBA2的下調和自噬的激活則被TMA7-OE拯救(圖8A)。在此,TMA7-OE挽救了IGF2BP3-KD誘導的細胞增殖、侵襲和遷移的減少。相反,TMA7-KD挽救了IGF2BP3-OE誘導的細胞增殖、侵襲和遷移的增加(圖8B-E)。
結果表明,TMA7-OE挽救了IGF2BP3-KD引起的細胞凋亡增加。此外,TMA7-KD還挽救了IGF2BP3-OE引起的細胞凋亡的減少(圖 8F)。自噬通量表明TMA7-OE減弱了IGF2BP3-KD誘導的自噬增加。相反,TMA7-KD增強了IGF2BP3-OE誘導的自噬的減少(圖8G)。
因此,我們得出IGF2BP3和TMA7之間的相互作用,且IGF2BP3通過TMA7調節LSCC中UBA2的表達和自噬。
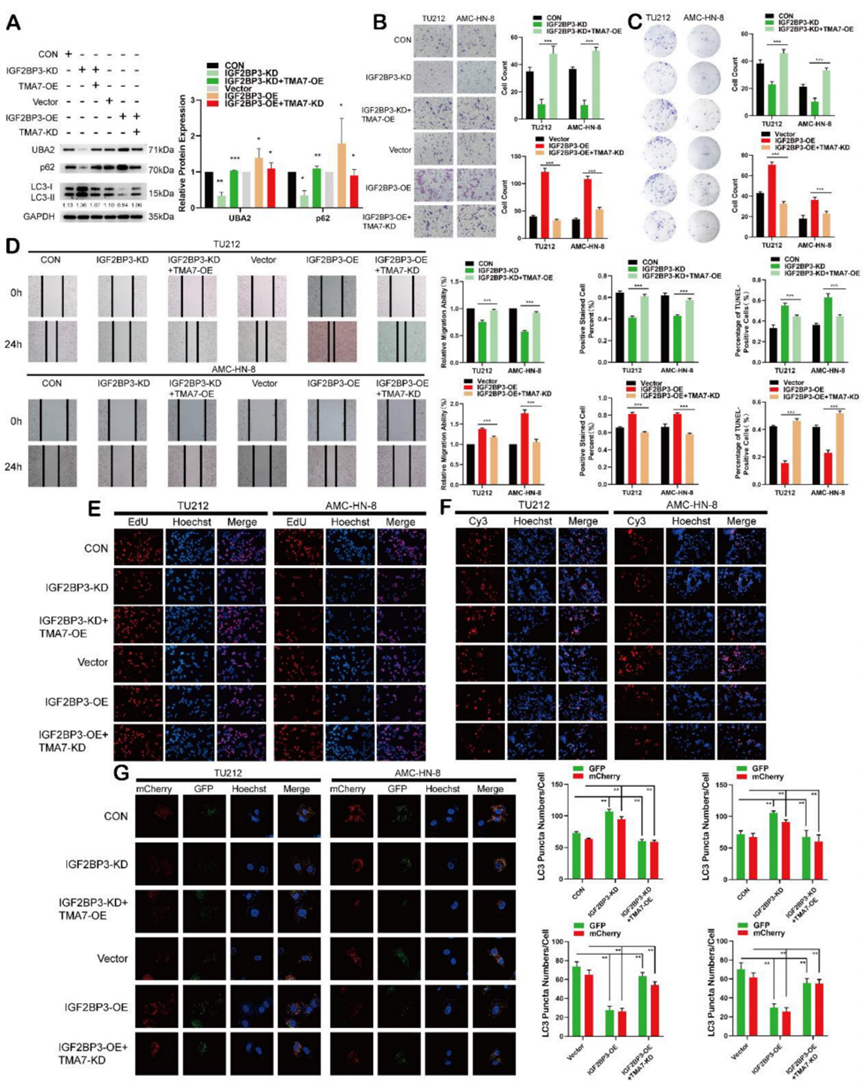 圖8 IGF2BP3通過TMA7調節LSCC自噬和UBA2表達
圖8 IGF2BP3通過TMA7調節LSCC自噬和UBA2表達
9、TMA7影響LSCC細胞自噬誘導的順鉑耐藥
為了檢查TMA7是否調節喉癌對順鉑的化療敏感性,我們構建了順鉑耐藥的AMC-HN-8/DDP和TU212/DDP細胞。結果顯示,TMA7增加可促進細胞活力并誘導順鉑處理細胞的化療耐藥性,而RAPA則使細胞對順鉑處理敏感。雷帕霉素可能會改變TMA7對LSCC細胞耐藥性的影響(圖9A-D)。RAPA降低了IC50,而TMA7-OE增加了IC50(圖9E)。
此外,我們替換了自噬抑制劑雷帕霉素,雷帕霉素可以抑制自噬并檢測自噬相關蛋白。Western blotting顯示TMA7-OE增加了p62并降低了LC3B-II/I比率,而雷帕霉素觸發了順鉑耐藥細胞的自噬(圖9F)。
接下來,我們檢查了TMA7對順鉑耐藥細胞中PI3K-mTOR信號傳導的影響。Western blot表明TMA7-OE增加,而雷帕霉素降低mTOR磷酸化水平。此外,TMA7-OE降低了BAX、cleaved-caspase3和cleaved-caspase9的水平,而雷帕霉素增加了這些蛋白的水平,表明由于順鉑耐藥細胞的凋亡水平較低(圖9G),TMA7-OE激活順鉑耐藥細胞中的PI3K-mTOR通路。
自噬通量表明雷帕霉素減弱了TMA7-OE誘導的自噬減少(圖9H)。在裸鼠體內,用TMA7-KD轉染的LSCC細胞(n=5)治療,另一組用TMA7-KD和UBA2-OE轉染的TU212穩定細胞治療(n=5),第三組注射TU212細胞。所有裸鼠飼養34天。異種移植數據顯示,低水平的TMA7顯著抑制異種移植瘤的生長,而UBA2-OE增強喉癌的致瘤性(圖9I,K,L)。
與未處理的LSCC細胞相比,TMA7-KD細胞中的自噬小體由線粒體結構組成,表明TMA7-KD激活了線粒體自噬并觸發了隨后的自噬(圖9J)。
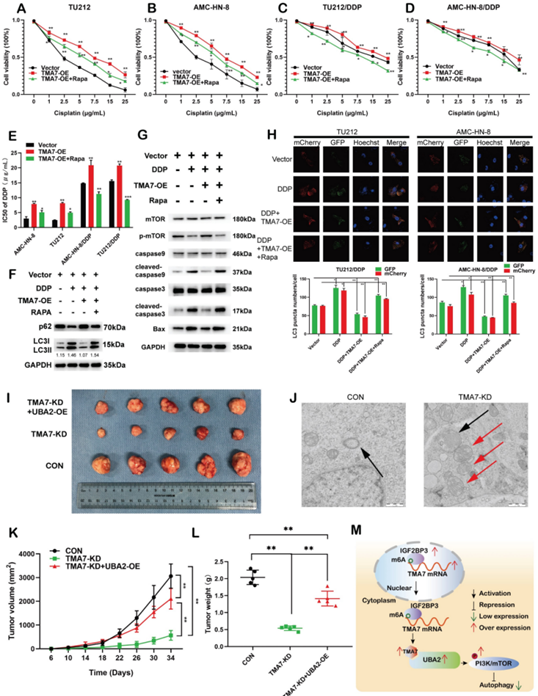
圖9 TMA7對自噬誘導LSCC細胞順鉑耐藥的影響
綜上所述,目前的研究表明,TMA7在喉鱗狀細胞癌中升高,與預后不良有關。TMA7是一種癌基因,調節喉癌細胞的增殖、侵襲、凋亡、自噬和化療敏感性。TMA7通過UBA2調節PI3K/mTOR通路,而IGF2BP3以m6A依賴的方式調節PI3K/mTOR通路。因此,這項研究可能為喉癌提供潛在的分子標記和治療靶點(圖9M)。
實驗方法
WB、轉錄組學和蛋白質組學多組學分析、集落形成試驗、Transwell侵襲試驗
參考文獻:
Yang L, Yan B, Qu L, Ren J, Li Q, Wang J, et al. IGF2BP3 Regulates TMA7-mediated Autophagy and Cisplatin Resistance in Laryngeal Cancer via m6A RNA Methylation. Int J Biol Sci. 2023;19(5):1382-400. Epub 2023/04/15. doi: 10.7150/ijbs.80921. PubMed PMID: 37056932; PubMed Central PMCID: PMCPMC10086756.