






m6A去甲基化酶FTO通過MCM3介導的細胞周期進展和HIF-1α激活穩定LINK-A發揮致癌作用

食管癌是一種高度惡性的腫瘤,2020年在全球范圍內發病率排名第七,死亡率排名第六。食管鱗狀細胞癌(ESCC)是食管癌最常見的組織學亞型,預后較差。化療是食管癌的一線治療,尤其是對于無手術指征的局部晚期食管癌患者。然而由于化療耐藥,食管癌患者的5年生存率不足20%。因此,探索關鍵的致癌驅動基因和闡明潛在的相關分子機制,以確定食管鱗癌有前景的治療靶點是值得的。N6-甲基腺苷(m6A)修飾是真核生物中最豐富的內部RNA修飾,主要發生于mRNA。非編碼RNA(ncRNA)的m6A修飾也在包括癌癥發生在內的各種生理和病理生物過程中發揮重要作用。新證據表明,m6A修飾可在不同情況下發揮致癌或腫瘤抑制作用。然而,在癌癥進展過程中,m6A修飾調控ncRNA的確切機制仍然非常模糊。缺氧誘導因子1(HIF-1)是一種氧感知轉錄因子。氧反應性HIF-1α亞基和組成性表達的HIF-1β亞基相互作用形成異二聚體HIF-1轉錄因子,在細胞對低氧的應答中起關鍵作用。HIF-1α的積累以及HIF-1α介導的葡萄糖轉運蛋白和葡萄糖代謝限速酶的激活降低了氧化磷酸化的效率,促進了糖酵解。雖然有報道稱HIF-1α的轉錄激活可導致癌癥代謝重編程和治療抵抗,但lncRNA在食管鱗癌中調節HIF-1α激活的機制仍不清楚。該研究發表在《Cell Reports》,IF:9.995。
技術路線

主要研究結果
1. FTO穩定LINK-A并與化療耐藥相關
為了研究在食管鱗癌化療耐藥背景下關鍵lncRNA的m6A修飾,研究者收集了34例接受新輔助化療的食管鱗癌患者的基線樣本(17例化療耐藥和17例化療敏感),然后測定了兩種主要的m6A去甲基化酶FTO和ALKBH5的mRNA表達水平。與化學敏感樣本相比,在化學耐藥樣本中顯著上調(圖1A,B)。此外,研究者測定了FTO在先前建立的親本和化療耐藥ESCC細胞系中的蛋白水平與相應親本細胞相比,FTO蛋白表達在兩個化學耐藥細胞系中上調(圖1C)。與化療耐藥細胞中FTO表達上調一致,m6A斑點雜交實驗結果顯示,與親本細胞相比,化療耐藥細胞中m6A的總體水平降低(圖1D)。隨后,為了進一步識別FTO調控的lncRNA,研究者在兩個食管鱗癌細胞系中沉默和過表達FTO,然后測量了14個注釋的ncRNA的表達水平,這些ncRNA先前在化療耐藥的食管鱗癌細胞系中顯著高于相應的親本細胞系4個ncRNA(TTLL3、LINC00663、PRSS30P、CETN4P)的表達低于實時熒光定量pcr的檢測限。在其余的10個ncRNA中,只有LINK-A在KYSE30和KYSE450細胞中在FTO敲低后顯著下調,在FTO過表達后顯著上調(圖1E,F)。
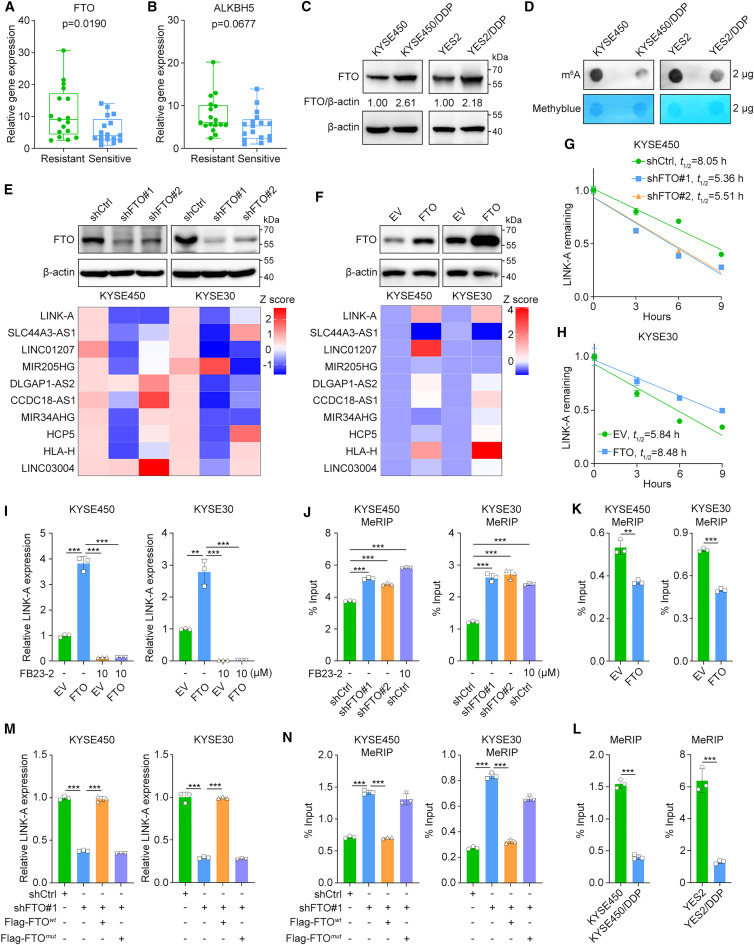
圖1 FTO穩定LINK-A并與化療耐藥相關
接下來,研究者進行了RNA穩定性分析,發現FTO敲低顯著縮短了LINK-A的半衰期,而FTO過表達穩定了LINK-A(圖1G,H)。隨后,研究者用FTO抑制劑FB23-2處理對照組和過表達FTO的KYSE30和KYSE450細胞。FTO過表達顯著增加了LINK-A的表達水平,而FB23-2處理則減弱了這一作用(圖1I)。此外,研究者還確定了FTO對LINK-A的穩定是否依賴于其去甲基化酶活性。甲基化RNA免疫沉淀(MeRIP)-qPCR結果表明,FTO敲低和FB23-2處理均顯著增加,但FTO過表達降低了LINK-A的m6A水平(圖1J,K)。與這一發現一致,MeRIP-qPCR結果顯示,與親本細胞相比,化療耐藥細胞中LINK-A的m6A水平顯著降低(圖1L)。實時qPCR結果表明,在KYSE30和KYSE450細胞中,野生型但未突變的FTO逆轉了FTO敲低引起的LINK-A表達下降(圖1M)。此外,MeRIP-qPCR結果表明,在FTO沉默的KYSE30和KYSE450細胞中,野生型FTO轉導導致LINK-A甲基化水平顯著降低,而這種去甲基化在FTO突變體轉導后未發生(圖1N)。這些結果表明FTO以m6a依賴的方式穩定LINK-A的表達。
2. LINK-A在食管鱗癌中具有致癌作用
為了研究LINK-A在食管鱗癌中的作用,研究者通過原位雜交分析了LINK-A在食管鱗癌患者隊列中的表達。生存分析表明,高LINK-A表達與不良預后強相關(圖2A)。此外,LINK-A在癌組織中的表達顯著高于正常組織(圖2B)。此外,在GEO數據庫中驗證了LINK-A在癌組織中的顯著上調(圖2C)。為了進一步評估LINK-A作為食管鱗癌治療靶點的潛力,研究者使用了體外化療耐藥模型。研究者測定了長期誘導化療耐藥的食管鱗癌細胞(KYSE450/DDP和YES2/DDP)和相應親本細胞(KYSE450和YES2)中的LINK-A表達水平,發現與親本細胞相比,化療耐藥細胞中的LINK-A表達顯著上調(2~5倍)(圖2D)。為了確定LINK-A在藥物短期暴露反應中的變化,研究者用順鉑(DDP)處理ESCC細胞不同時間(0、6、12和24 h)。隨著DDP處理時間的增加,LINK-A的表達顯著增加,尤其是在處理24 h后(圖2E)。這些結果表明,LINK-A可能在化療耐藥的獲得和維持中起著至關重要的作用。
最后,異種移植實驗表明,在KYSE450/DDP和KYSE450細胞中沉默LINK-A顯著延緩了腫瘤生長并誘導了化療敏感性,而在KYSE150細胞中過表達LINK-A顯著促進了腫瘤生長和對細胞毒性化療的耐藥性(圖2F-K)。綜上所述,這些結果表明,LINK-A在食管鱗癌中起著致癌作用。

圖2 LINK-A在食管鱗癌中發揮致癌作用
3. LINK-A通過促進MCM3與CDK1的相互作用促進MCM3磷酸化
為了進一步闡明LINK-A致癌的下游調控機制,研究者通過質譜分析鑒定了KYSE450和YES2細胞中LINK-A沉淀的蛋白質。在KYSE450和YES2細胞中分別鑒定出43和14種LINK-A相互作用蛋白,并且在兩個細胞系中均鑒定出其中7種相互作用蛋白(圖3A)。在這7個重疊蛋白中,MCM3已被報道在細胞周期進程和DNA復制中發揮重要作用,但其在食管鱗癌中的詳細分子機制,特別是介導癌細胞增殖和化療耐藥的機制尚不清楚。接下來,研究者進行了RNA pull-down和蛋白質印跡,以確認MCM3和LINK-A的義鏈而不是反義鏈之間的特異性相互作用(圖3B)。此外,研究者通過RIP-qPCR在KYSE30和KYSE450細胞中驗證了這種相互作用(圖3C)。接下來,研究者探究了LINK-A對其相互作用蛋白MCM3表達的影響。敲低或過表達LINK-A均不影響MCM3的mRNA或蛋白水平(圖3D,E)。值得注意的是,蛋白質印跡分析表明,LINK-A顯著促進MCM3在Ser112的磷酸化(圖3D,E)。此外,在瞬時LINK-A過表達后,MCM3 Ser112的磷酸化水平以劑量依賴的方式增加(圖3F,G)。
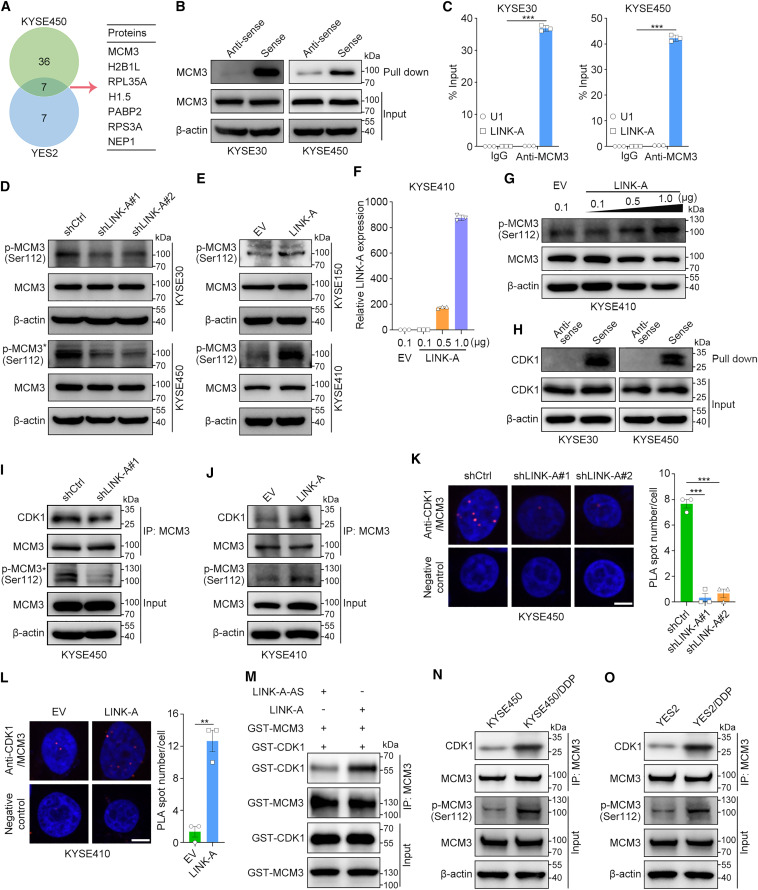
圖3 LINK-A通過促進MCM3與CDK1的相互作用促進MCM3磷酸化
先前的研究報道,CDK1在112位絲氨酸磷酸化MCM3,并介導MCM2-7復合物的組裝和染色質裝載然后研究者確定了LINK-A是否通過作為CDK1和MCM3的支架來促進MCM3的磷酸化。RNA pull down和隨后的蛋白質印跡結果證實了KYSE30和KYSE450細胞中LINK-A和CDK1之間的相互作用(圖3H)。接下來,免疫共沉淀(coIP)實驗的結果表明,敲低LINK-A顯著抑制和過表達LINK-A增加了CDK1和MCM3之間的相互作用(圖3I,J)。鄰近連接實驗結果證實,在體內,LINK-A促進了CDK1和MCM3之間的相互作用(圖3K,L)。此外,研究者使用純化的MCM3和CDK1蛋白在含有LINK-A有義或反義鏈的緩沖液中進行了體外pull-down實驗。結果表明,LINK-A有義鏈而非反義鏈的存在促進了MCM3和CDK1的體外相互作用(圖3M)。與此一致的是,與相應的親本細胞系相比,MCM3在Ser112的磷酸化以及CDK1和MCM3之間的相互作用在兩種耐藥細胞系中都增加了(圖3N,O)。
4. 磷酸化MCM3介導了LINK-A的致癌作用
為了確定LINK-A介導的MCM3磷酸化是否促進MCM復合物的組裝和隨后的染色質加載,研究者進行了細胞分離,然后通過蛋白質印跡分析檢測染色質結合蛋白。在ESCC細胞中,敲低LINK-A顯著降低MCM2、MCM3、MCM4、MCM5、MCM6和MCM7蛋白的染色質負荷,而過表達LINK-A促進MCM2-7復合體的染色質負荷(圖4A,B)。接下來,研究者構建了MCM3的Ser112位點突變體(MCM3S112A;Ser112突變為丙氨酸),并將野生型MCM3和MCM3S112A轉染至去除內源性MCM3的細胞中。由LINK-A介導的MCM2-7復合物的染色質負荷增加被Ser112突變所消除,表明這一過程依賴于MCM3的磷酸化(圖4C)。為了確定MCM3是否介導了LINK-A對細胞周期進展的促進作用,研究者進行了挽救試驗,發現野生型MCM3的過表達完全挽救了LINK-A沉默介導的G0/G1期阻滯,而MCM3S112A沒有(圖4D)。相反,在LINK-A過表達的KYSE410細胞中沉默MCM3可抑制LINK-A介導的細胞周期進展(圖4E)。此外,IncuCyte活細胞成像分析表明,野生型MCM3而不是MCM3S112A介導了LINK-A對細胞增殖的促進作用(圖4F,G)。為了進一步證明體內MCM3和LINK-A之間的功能關系,研究者使用KYSE150細胞進行了異種移植實驗。過表達LINK-A顯著促進細胞增殖和化療耐藥性,而沉默MCM3則消除了這些作用(圖4H,I)。綜上所述,這些結果表明,LINK-A通過增加MCM3磷酸化來促進MCM2-7復合體的組裝和隨后的染色質加載,從而發揮致癌作用。

圖4 磷酸化的MCM3介導了LINK-A的致癌作用
5. LINK-A通過從MCM3中分離HIF-1α來誘導HIF-1α轉錄活性
為了進一步研究LINK-A/MCM3軸導致細胞毒性化療耐藥性的機制,研究者在LINK-A沉默的KYSE450細胞中進行了RNA測序分析。引人注目的是,基因集富集分析表明,與由兩個獨立的短發夾RNA序列中的任何一個介導的LINK-A敲低的細胞相比,糖酵解途徑在對照細胞中顯著富集(圖5A)。鑒于之前一項研究觀察到MCM3通過直接與HIF-1α相互作用負調控HIF-1α轉錄活性,研究者試圖確定LINK-A是否激活HIF-1α以促進腫瘤糖酵解。首先,研究者在常氧條件下,通過在LINK-A沉默的KYSE450細胞和LINK-A過表達的KYSE410細胞中進行IP實驗,確定LINK-A是否影響MCM3和HIF-1α的相互作用。結果表明,沉默LINK-A顯著增加MCM3與HIF-1α的相互作用,而過表達LINK-A則抑制MCM3與HIF-1α的相互作用(圖5B)。此外,研究者通過GST pull-down純化了MCM3和HIF-1α蛋白,然后在含有LINK-A的有義或反義鏈的緩沖液中進行了體外coIP實驗。在體外,LINK-A有義鏈而非反義鏈的存在抑制了MCM3和HIF-1α之間的相互作用(圖5C)。沉默LINK-A顯著抑制了缺氧反應元件驅動的熒光素酶活性,而過表達LINK-A增強了低氧反應元件驅動的熒光素酶活性(圖5D,E)。此外,實時熒光定量PCR結果表明,在常氧和低氧條件下,LINK-A顯著促進了HIF-1α靶基因的表達,包括SLC2A1、PDK1、HK2、LDHA和血管內皮生長因子(圖5F,G)。值得注意的是,與常氧條件相比,在低氧條件下,LINK-A和HIF-1α靶基因的表達顯著上調(圖5F,G),進一步證實了LINK-A和HIF-1α轉錄活性之間的功能相關性。
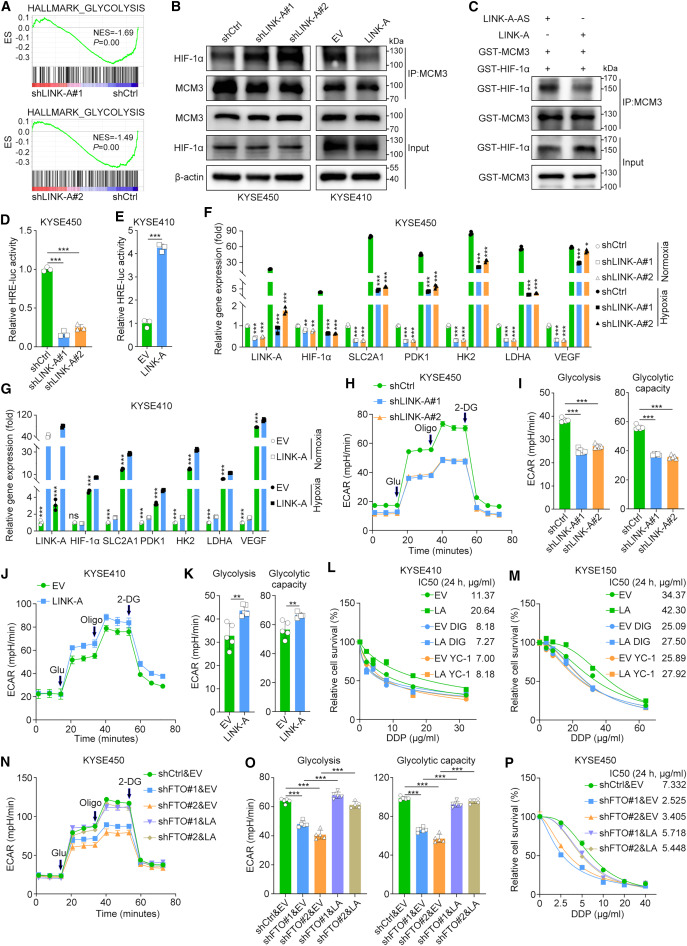
圖5 LINK-A通過將HIF-1α從MCM3中分離來誘導HIF-1α轉錄激活
鑒于HIF-1α在癌癥代謝重編程中的重要作用,研究者進行了Seahorse實驗來確定LINK-A對腫瘤糖酵解的影響。敲低LINK-A顯著降低了細胞外酸化率(ECAR),但適度增加了氧消耗率(OCR)(圖5H,I)。同樣,LINK-A過表達增加了ECAR,但降低了OCR(圖5J,K)。進一步研究HIF-1α是否介導LINK-A在食管鱗癌化療耐藥中的作用。細胞活力測定結果表明,HIF-1α抑制劑lificiguat(YC-1)和Digoxin處理顯著抑制了KYSE410和KYSE150細胞中LINK-A的致癌作用(圖5L,M)。綜上所述,這些結果表明,LINK-A通過消除MCM3介導的HIF-1α轉錄抑制來激活HIF-1α,進而促進糖酵解和化療耐藥。Seahorse檢測的結果顯示,FTO敲低后糖酵解活性顯著降低,隨后在LINK-A過表達后觀察到這種降低的逆轉(圖5N,O)。與這一發現一致,在FTO沉默細胞中過表達LINK-A恢復了增殖減少和化療耐藥性降低的表型(圖5P)。
6. 靶向LINK-A可使食管鱗癌對細胞毒性化療增敏
為了確定靶向LINK-A是否為食管鱗癌有前景的治療策略,研究者建立了3個PDX模型。腺相關病毒介導的LINK-A沉默顯著增加了ESCC pdx對DDP的敏感性(圖6A-F)。最后,研究者在圖1A所示的相同基線樣本中測定了LINK-A的轉錄水平。結果表明,與化療敏感樣本相比,化療耐藥樣本中LINK-A的表達水平顯著上調(圖6G)。進一步的相關性分析顯示,在這些樣本中,LINK-A的表達與FTO的表達呈正相關(圖6H),進一步證實了在ESCC化療耐藥過程中FTO對LINK-A的調控作用。綜上所述,研究者的研究結果證實了LINK-A是食管鱗癌化療耐藥的罪魁禍首,揭示了LINK-A介導食管鱗癌化療耐藥的潛在機制以及LINK-A在食管鱗癌化療耐藥中的穩定性調節。
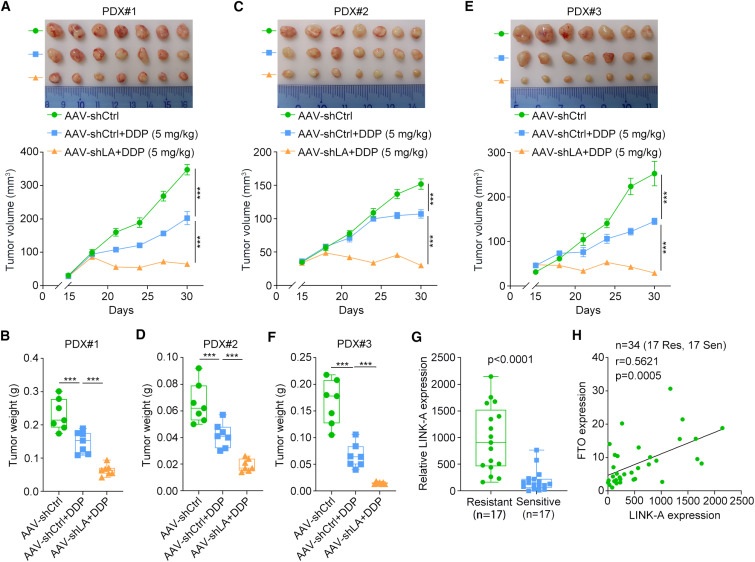
圖6 靶向LINK-A使食管鱗癌對細胞毒性化療增敏
結論
該研究揭示了食管鱗癌細胞中LINK-A的m6A去甲基化的細節。m6A去甲基化酶FTO介導了LINK-A的穩定并支持其致癌作用。LINK-A與MCM3之間的直接相互作用不僅增加了CDK1介導的MCM3 Ser112的磷酸化,從而促進MCM2-7復合體的組裝和染色質負載,以及隨后的細胞周期進展和細胞增殖,而且還從MCM3介導的轉錄抑制中釋放HIF-1α,從而促進腫瘤代謝重編程和化療耐藥。綜上所述,該研究結果提示FTO/LINK-A/MCM3/HIF-1α軸在食管鱗癌的進展中起著至關重要的作用,這一發現為靶向LINK-A在食管鱗癌患者,尤其是LINK-A高表達患者中的治療提供了潛在的策略。
機制圖

實驗方法
慢病毒的產生和感染,細胞活力測定,活細胞成像,細胞周期分析,蛋白質印跡,RT-qPCR,m6斑點印跡測定,RNA pull-down,RIP,mRNA穩定性測定,雙熒光素酶報告基因檢測,FISH,IF染色,海馬檢測,RNA-seq和數據分析,小鼠實驗
參考文獻
Nan Y, Liu S, Luo Q, Wu X, Zhao P, Chang W, Zhang R, Li Y, Liu Z. m6A demethylase FTO stabilizes LINK-A to exert oncogenic roles via MCM3-mediated cell-cycle progression and HIF-1α activation. Cell Rep. 2023 Oct 18;42(10):113273. doi: 10.1016/j.celrep.2023.113273.