






YY1液滴復合物澆灌M2巨噬極化導致前列腺癌進展
腫瘤相關巨噬細胞主要是M2極化表型,通過分泌多種細胞因子重塑腫瘤微環境,促進腫瘤進展。本研究通過過表達YY1的轉基因小鼠證明了YY1在M2巨噬細胞中的作用,并研究了靶向M2巨噬細胞YY1治療前列腺癌(PCa)的潛力。此外,YY1通過調節相分離和增強子-啟動子相互作用,從而促進巨噬細胞中IL- 6的轉錄、從而重塑腫瘤微環境。本文于2023年4月發表在《Journal of Immunotherapy of Cancer》IF: 11.4期刊上。
技術路線

主要實驗結果
1、在體內YY1過表達增加M2巨噬細胞浸潤和PCa進展
從oe-YY1轉基因小鼠和野生型小鼠中提取腹腔源性巨噬細胞(PCDM)和骨髓源性巨噬細胞(BMDM)。流式細胞術發現,oe- YY1小鼠巨噬細胞中CD163+ M2的比例明顯增加,而CD86 + M1的比例沒有明顯增加(圖1A-C)。此外,oe- YY1小鼠的PCa腫瘤負荷比野生型生長更快(圖1D)。免疫熒光顯示離體腫瘤中CD163+ M2的顯著更高其oe- YY1小鼠顯著多于野生小鼠(圖1E)。在oe- YY1小鼠中還觀察到低豐度的CD4和CD8細胞的浸潤(圖1F)。流式細胞術證實oe- YY1小鼠腫瘤中M2細胞比較更高,CD8毒性T細胞比例更低(圖1G),且伴隨更低的IFNγ的表達(圖1H),說明過表達YY1降低T細胞的抗腫瘤活性。隨后構建肺轉移模型,結果顯示過表達YY1帶來的腫瘤生成效果被巨噬細胞清除劑clodronate liposomes所抑制(圖1I)。將oe- YY1小鼠來源的骨髓細胞注射至野生小鼠構建嵌合小鼠,然后皮下成瘤,結果顯示,與對照組比較,YY1過表達組小鼠的腫瘤更大(圖1J)。
隨后將M2巨噬細胞靶向肽(M2pep)合成至脂質體載體上,其轉染了YY1 siRNA(M2pep-siYY1)用于靶向M2巨噬細胞的YY1。M2pep-siYY1和對照siNC尾靜脈注射至oe- YY1小鼠PCa肺轉移小鼠中。結果顯示M2pep-siYY1組的存活顯著延長(圖1K),PD-1抑制劑聯合處理進一步顯著延長小鼠存活并減少轉移結節(圖1L)。HE和IHC染色顯示M2pep-siYY1和PD-1處理顯著降低肺結節并增加CD8和CD4T細胞的浸潤(圖1M)。總之,以上結果表明在體內YY1過表達增加M2巨噬細胞浸潤和PCa進展。

圖1在體內YY1過表達增加M2巨噬細胞浸潤和PCa進展
2、YY1在人前列腺癌中與CD163 + M2巨噬細胞呈正相關
為驗證YY1和巨噬細胞的關系,作者利用IHC檢測了臨床隊列PCa和癌旁樣本中YY1和CD163的表達。有趣的是,作者發現YY1不僅在惡性細胞上表達,而且在CD163 +巨噬細胞浸潤的腫瘤基質中,特別是在巨噬細胞樣細胞中,也表現出相當的表達水平(圖2A)。多色IHC顯示,YY1在CD163巨噬細胞浸潤的腫瘤機制中表達(圖2B-C)。IHC組織芯片結果顯示YY1得分在腫瘤基質和轉移淋巴結中都顯著高于正常對照(圖2D-E)。此外,YY1得分越高的腫瘤基質中CD163的表達越高(圖2F)。YY1得分高于不良預后相關(圖2G),而YY1和CD163得分都高的患者預后也更差(圖2H)。綜上所述,通過對臨床組織的分析,我們發現YY1在前列腺腫瘤浸潤的M2巨噬細胞上表達,并且腫瘤基質中YY1的表達也與CD163 +巨噬細胞的含量呈正相關。
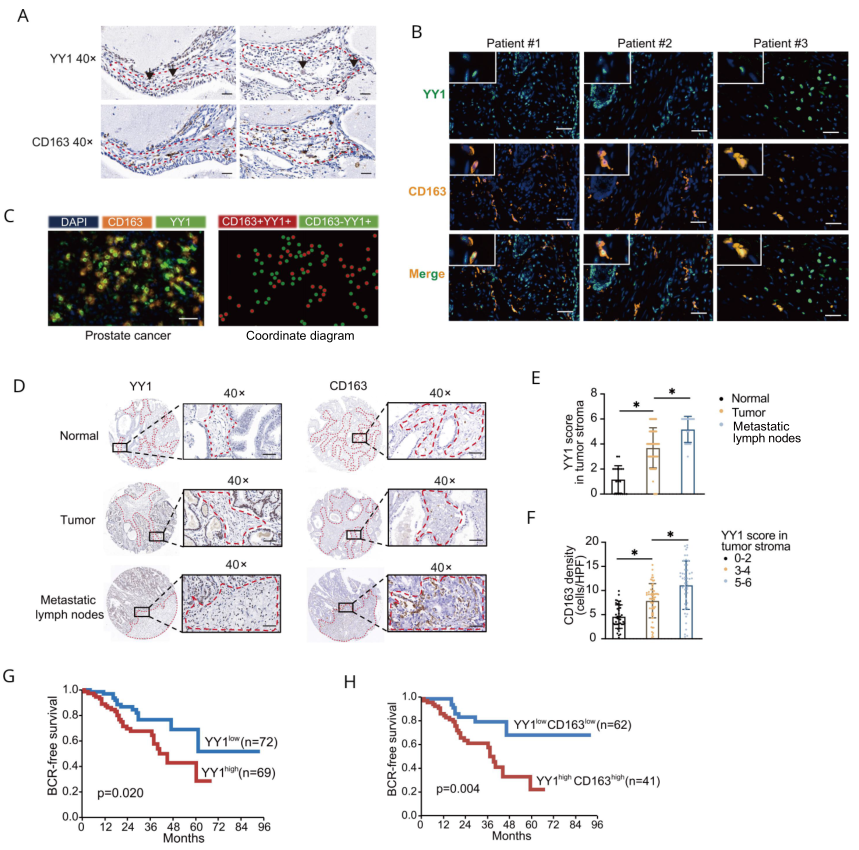
圖2 YY1在人前列腺癌中與CD163 + M2巨噬細胞呈正相關
3、YY1通過IL- 4/STAT6通路上調,參與M2巨噬細胞極化
鑒于上述結果,作者開始檢測YY1是否調控巨噬細胞M2極化。利用THP-1誘導M0和M2巨噬細胞(圖3A)。流式細胞術證實了CD206標記的M2巨噬細胞的比例(圖3B)。巨噬細胞M2極化也被qRT- PCR和WB證實,且YY1的表達在M2巨噬細胞中顯著高于M0巨噬細胞(圖3C-D)。因此,成功誘導M2巨噬細胞且YY1在其內表達。
有趣的是,作者觀察到YY1過表達誘導的細胞形態變化和M0誘導M2極化的變化類似(圖3E-F)。此外,YY1過表達顯著促進M2相關標志物的表達抑制M1標志物表達(圖3G),并顯著促進M2巨噬細胞的比例(圖3H)。這些結果表明YY1促進M2巨噬細胞極化。數據庫預測分析發現STAT6是YY1啟動子結合的參與M2極化調控IL-4信號通路的關鍵轉錄因子。雙熒光素酶和ChIP實驗證實了該結果(圖3I-J)。IL-4誘導的M2巨噬細胞中STAT6高度磷酸化。綜上,YY1通過IL- 4/STAT6通路上調,參與M2巨噬細胞極化。

圖3 YY1通過IL- 4/STAT6通路上調,參與M2巨噬細胞極化
4、YY1增加M2巨噬細胞誘導的前列腺癌惡性表型
通過使用M0和M2巨噬細胞的條件培養基(CM)共培養PCa細胞系證實了M2巨噬細胞的促腫瘤作用(圖4A)。隨后,利用YY1過表達或抑制的M2巨噬細胞CM孵育PCa細胞,結果顯示與對照組比較,YY1過表達M2的CM顯著促進細胞的遷移,增殖和集落形成,敲低則相反(圖4B-F)。與oe- YY1 PBMC來源的巨噬細胞CM培養14天后,PCa患者的人原發性癌細胞類器官的平均半徑和面積也有所增加(圖4G)。與oe- YY1 M2 RAW264.7細胞混合的皮下成瘤模型腫瘤的體積和重量都顯著增加(圖4H),肺轉移結節也顯著增多(圖4I)。隨后,對oe- YY1 M2巨噬細胞CM或對照CM處理的LNCaP細胞進行RNA測序,發現實驗組上調的基因主要富集至NF-κB和JAK- STAT信號通路(圖4J-K),且WB證實了STAT3和p65的高表達。
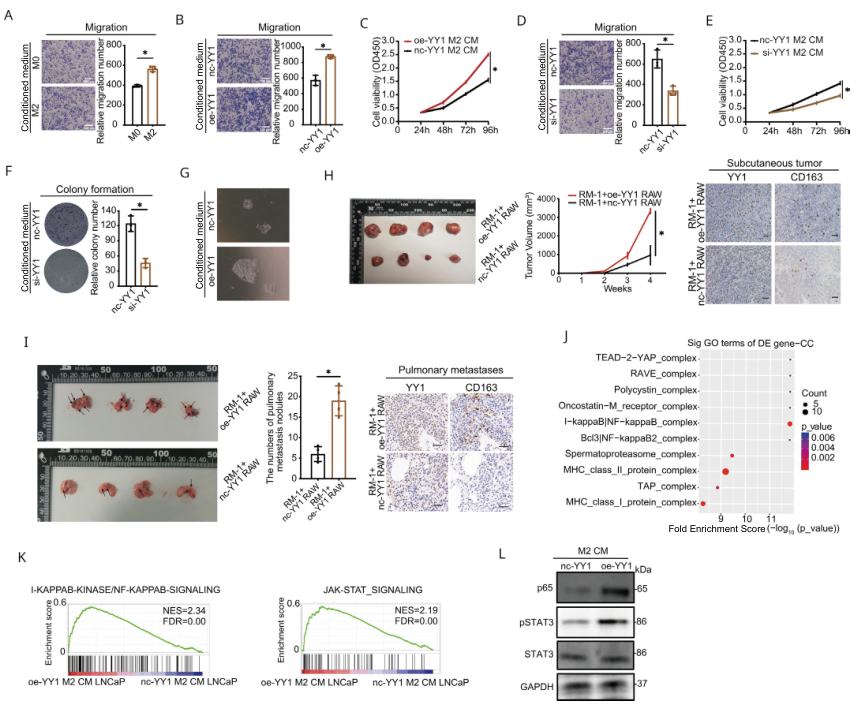
圖4 YY1增加M2巨噬細胞誘導的前列腺癌惡性表型
5、M2巨噬細胞中YY1上調IL- 6可增加前列腺癌惡性表型
作者在oe- YY1和si- YY1 M2巨噬細胞CM以及nc- YY1 M2 CM中進行了27種與腫瘤免疫相關的細胞因子液懸芯片檢測。與對照組相比,oe- YY1組M2巨噬細胞CM中IL- 6水平顯著上調,YY1抑制組IL- 6水平顯著下調(圖5A-B)。WB證實YY1過表達促進IL-6的表達,敲除YY1則相反(圖5C-E)。因此,接下來探究IL-6是否是YY1誘導促腫瘤的關鍵,進行了回復實驗。結果顯示過表達IL-6(rIL-6)顯著促進因YY1敲除引起的腫瘤抑制(圖5F-I),相反加IL-6抗體沉淀IL-6則顯著逆轉YY1過表達對腫瘤惡性表型的影響(圖5J-M)。這些結果表明YY1通過IL-6促進PCa的惡性表型。
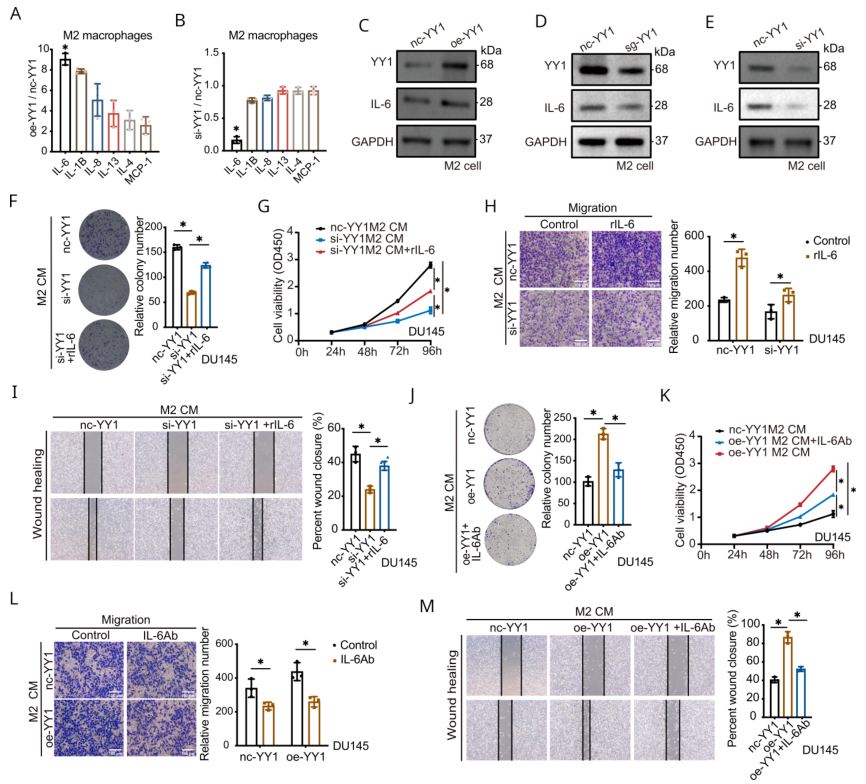
圖5 M2巨噬細胞中YY1上調IL- 6可增加前列腺癌惡性表型
6、YY1通過調節p300、p65和CEBPB來調節IL- 6的表達
為進一步研究YY1在巨噬細胞中調控IL- 6的詳細機制,根據JASPAR數據庫預測了IL- 6啟動子區域的主要轉錄因子。有趣的是,在IL- 6啟動子區域發現了四個YY1結合位點(P1-P4),還發現YY1、CEBPB和p65的基序位于IL- 6啟動子附近(圖6A)。使用生物信息學分析了這4個蛋白的互作,發現YY1與其它三個蛋白結合并處于中心地位(圖6B)。ChIP-qPCR結果證實YY1、CEBPB和p65都結合至IL-6的啟動子區域(圖6C-E)。ChIP- on- ChIP實驗證實4個蛋白都結合在IL-6啟動子的P4區域(圖6F)。雙熒光素酶實驗也證實了YY1、CEBPB和p65都結合IL-6的啟動子區域并增強該區域的活性(圖6G-J)。這些結果表明YY1、p300、p65和CEBPB作為上游轉錄因子,通過結合IL- 6啟動子區域促進IL- 6轉錄。
從M2巨噬細胞中提取總蛋白,對YY1、p65、CEBPB和p300進行共免疫沉淀和免疫印跡,以驗證它們之間的相互作用(圖6K-M)。p300基因敲除或復合物抑制劑金絲桃素可抑制它們之間的相互作用(圖6N、O)。此外,金絲桃素不僅減少了M2巨噬細胞中IL- 6 mRNA的表達(圖6P),還降低了與M2巨噬細胞CM孵育的PCa細胞的增殖和遷移(圖6Q、R)。
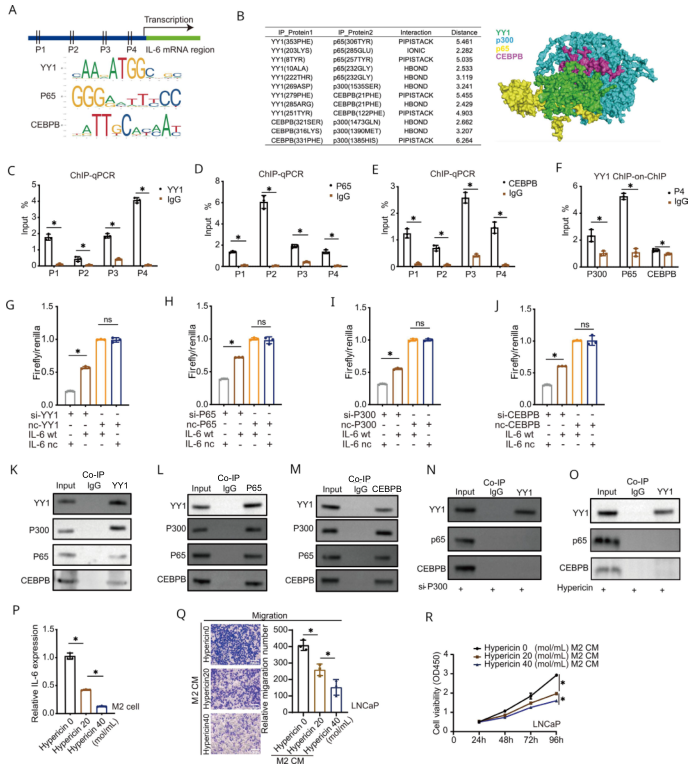
圖6 YY1通過調節p300、p65和CEBPB來調節IL- 6的表達
7、YY1通過M2特異性增強子促進IL- 6轉錄
作者對PCa的腫瘤組織和癌旁組織的單細胞測序結果進行分析,發現與前列腺正常組織巨噬細胞相比,YY1和輔因子CEBPB在PCa巨噬細胞中轉錄活性均顯著升高,均排在前20位轉錄因子列表(圖7A)。對M2巨噬細胞和THP- 1細胞進行了H3K27ac和YY1的ChIP- seq,發現了數千個在M2極化過程中發生變化的獲得或丟失的增強子(圖7B)。在M2極化過程中,共有15241個增強子被鑒定為M2特異性增強子。THP- 1細胞中M2特異性增強子的H3K27ac信號較低,M2巨噬細胞中M2特異性增強子的YY1 ChIP- seq信號也富集(圖7C)。無論如何,M2特異性增強子的YY1 ChIP- seq信號顯著下降,表明YY1在M2巨噬細胞中發揮重要作用。
為鑒定IL- 6的不同增強子,使用Hi-C數據鑒定增強子-啟動子相互作用。發現M2特異性IL-6增強子與IL- 6啟動子直接相互作用(圖7B,D),并且在其周圍有明顯更強的H3K27ac和YY1ChIP- seq 信號在極化的M2巨噬細胞中,與THP- 1細胞比較(圖7C)。為證實位于Hi- C數據中的IL- 6增強子的作用,構建一個熒光素酶基因質粒,其中含有四個片段(E1-4區)的增強子片段(圖 7E)。然后和過表達YY1載體共轉染到M2巨噬細胞中,通過熒光素酶檢測和染色體構象捕獲(3C)實驗,確定了E1區(增強子片段的遠端)是YY1的結合位點(圖7F-G)。用CRISPR- Cas9敲除M2巨噬細胞中的E1區(sg-E1),ELISA和qRT- PCR顯示IL- 6表達量減少(圖7H-I)。溴域抑制劑JQ1抑制了增強子信號也下調了IL- 6的表達(圖7J)。這些結果表明這些結果表明,M2-特異性IL-6增強子對IL- 6轉錄的重要作用。
接下來,進行體外和體內實驗,以驗證M2特異性增強子對M2巨噬細胞極化和功能的影響。首先,經JQ1處理的巨噬細胞中ARG-1和IL-10的mRNA表達量較低,表明JQ1抑制了M2的極化(圖7K)。此外,當在DU145細胞和M2巨噬細胞的共培養體系中加入JQ1時,與陰性對照相比,惡性細胞的遷移和侵襲能力明顯受到抑制(圖7L)。為精確檢驗M2特異性增強子對M2巨噬細胞功能的影響,用sg- E1 M2 CM培養DU145細胞。在sg- E1組中,PCa細胞的惡性行為降低(圖7M)。在體內實驗中,小鼠皮下注射腫瘤2周后,連續腹腔注射JQ1和CPI- 637(分別是針對BRD4和p300/CBP溴鏈的增強子抑制劑)3周后再收獲腫瘤,發現腫瘤體積與陰性對照組相比明顯縮小(圖7N)。總之,YY1復合物通過M2-特異性增強子上調IL-6,從而促進PCa的進展。
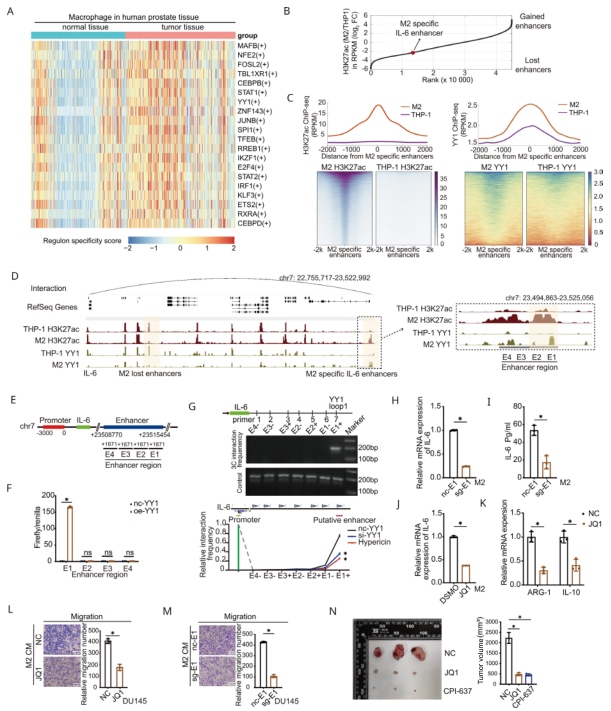
圖7 YY1通過M2特異性增強子促進IL- 6轉錄
8、YY1在M2巨噬細胞極化過程中形成LLPS
對YY1進行免疫熒光時,IL- 4刺激的M2巨噬細胞核內液滴數量較PMA- THP- 1 M0細胞明顯增加(圖8A),表明YY1可能通過LLPS形成凝析油。接下來,根據PONDR評分預測YY1的大IDRs(圖8B)。通過熒光顯微鏡,觀察到LLPS液滴的密度隨YY1- IDR- EGFP蛋白濃度的增加而增加(圖8C),但在使用1,6- 己二醇(1,6- Hex)處理后,液滴密度下降,因為1,6- Hex 會破壞介質中的弱疏水鍵。在YY1-非-IDR-EGFP細胞中檢測不到液滴(圖8C)。
接著,將YY1- IDR- EGFP、YY1-非 IDR-EGFP 和EGFP標記的YY1全長質粒轉染到M2巨噬細胞中,并在共聚焦熒光顯微鏡下觀察活細胞。與YY1-非IDR- EGFP組相比,YY1- EGFP組和YY1-IDR-EGFP組的液滴數量明顯增加(圖8D)。此外,還在一些液滴中觀察到融合和裂解(圖8E),并且在光漂白目標病灶后,核穿刺點的熒光強度可以恢復(圖8F),這表明巨噬細胞中的核凝聚物是由LLPS形成的。為進一步明確YY1液滴在巨噬細胞中的位置以及IDR區在液滴形成中的作用,在細胞固定后染色DAPI,并通過全長YY1 CRISPR/Cas9敲除YY1-IDR區,以及在M2巨噬細胞中過表達非IDR區(sg- YY1- IDR)。發現YY1液滴主要位于M2巨噬細胞核內,YY1- IDR敲除后液滴數量顯著減少(圖8G,H)。
此外,YY1/p300/p65/CEBPB 復合物中預測的YY1結合位點位于YY1- IDR 段(圖6B)。用EGFP抗體進行免疫沉淀,然后進行免疫印跡,結果表明在M2巨噬細胞中p65、p300和CEBPB與YY1- IDR- EGFP相互作用(圖8I)。還觀察到YY1的LLPS凝聚物與p300、p65和CEBPB的共定位(圖8J)。同時,在抑制p300、p65和CEBPB表達的siRNA以及p300/p65復合物抑制劑金絲桃素的干預下,M2巨噬細胞中的YY1凝聚物被化解(圖8K)。簡而言之,這些結果表明YY1- IDR區段在YY1 LLPS中的關鍵作用以及YY1/p300/p65/CEBPB復合物促進了在M2巨噬細胞中形成YY1介導的LLPS。此外,與陰性對照相比,用1,6-Hex預處理或轉染sg-YY1-IDR后,IL- 4-刺激的巨噬細胞中M2標記(ARG-1和IL-10)和IL- 6的相對表達明顯降低(圖8L、M)。此外,敲除YY1復合體中的輔因子也降低了ARG- 1和IL- 10的表達(圖8N)。總之,YY1復合物在M2極化過程中形成了LLPS并且YY1介導的LLPS上調了M2巨噬細胞中IL-6的表達。

圖8在M2巨噬細胞極化過程中,YY1形成液-液相分離
實驗方法
臨床組織樣本采集,細胞培養和轉染,CRISPR-Cas9敲除技術,RT-qPCR,流式細胞術,WB,熒光素酶實驗,ChIP,ELISA,RNA測序,細胞增殖實驗,Transwell實驗,小鼠成瘤實驗,轉基因小鼠構建,單細胞測序,相分離液滴實驗,光漂白后熒光恢復(FRAP)試驗
參考文獻
Chen S, Lu K, Hou Y, You Z, Shu C, Wei X, Wu T, Shi N, Zhang G, Wu J, Chen S, Zhang L, Li W, Zhang D, Ju S, Chen M, Xu B. YY1 complex in M2 macrophage promotes prostate cancer progression by upregulating IL-6. J Immunother Cancer. 2023 Apr;11(4):e006020. doi: 10.1136/jitc-2022-006020.