






URI重編程p53野生型肝癌脂質代謝,減輕TKIs誘導的鐵死亡!
基于酪氨酸激酶抑制劑(TKIs)的全身治療晚期肝細胞癌(HCC)的臨床獲益受到耐藥性的限制。在這里,作者發現由非傳統的前折疊蛋白RPB5相互作用因子(URI)介導的脂質代謝重編程使HCC對TKIs誘導的鐵死亡具有抗性。在機制上,URI直接與TRIM28相互作用,并以依賴TRIM28- MDM2的方式促進p53泛素化和降解。重要的是,p53結合硬脂酰輔酶A去飽和酶1(SCD1)啟動子并抑制其轉錄。URI高表達與SCD1高表達相關,二者協同表達預示HCC預后不良和TKIs耐藥。SCD1抑制劑aramchol與氘化索拉非尼衍生物多納非尼聯合在p53-野生型HCC患者來源的類器官和移植腫瘤中顯示出良好的抗腫瘤作用。這種聯合治療對于具有野生型p53和高水平URI/SCD1的晚期HCC患者具有潛在的臨床益處。本文于2023年10月發表于《Nature Communication》,IF:16.6,Q1。
技術路線

主要實驗結果
1、URI促進肝癌細胞對TKIs誘導的鐵死亡的抵抗
首先,作者進行了RNA測序(RNA-seq)分析,以確定激活URI依賴性轉錄組改變。作者發現在shRNA(Ctrl)和shURI HepG2細胞之間共有1676個基因差異表達。使用KEGG途徑進行的功能富集分析顯示,鐵死亡是URI調控的最重要的改變途徑之一(圖1a)。基因集富集分析(GSEA)顯示,與對照組相比,shURI細胞中鐵死亡而不是凋亡信號呈陽性富集(圖1b)。這些結果提示URI可能在鐵死亡中起重要作用。同時,包括EGFR酪氨酸激酶抑制劑耐藥、脂解調節、p53信號通路和癌癥中的轉錄失調等途徑也受到URI的調節(圖1a)。
有許多臨床試驗評估TKIs治療晚期HCC。與細胞毒性作用一致,這些TKIs在測試的各種癌細胞系中有效地降低了腫瘤活力(圖1c)。值得注意的是,以B-RAF為共同靶點的索拉非尼、瑞格拉非尼和多納非尼有利于鐵中毒樣細胞死亡,而其他測試藥物對細胞脂質過氧化的影響很小(圖1d)。
然后,作者測試了URI是否調節TKIs誘導的細胞毒性。增加索拉非尼濃度處理JHH1和HepG2細胞48小時,觀察細胞增殖情況。URI的消耗顯著增加了JHH1和HepG2細胞對索拉非尼的敏感性,在JHH1細胞中,IC50(導致50%的活力抑制)從8.324 μM(Ctrl)降低到6.480 μM(shURI),在HepG2細胞中,分別從8.047 μM(Ctrl)降低到4.069 μM(shURI)(圖1e)。克隆形成實驗證實了URI在索拉非尼耐藥中的作用(圖1f, g)。有趣的是,在JHH1和HepG2細胞中,URI敲低顯著增加了索拉非尼誘導的脂質過氧化(圖1h),這表明URI可能抑制TKIs誘導的鐵死亡。此外,索拉非尼誘導的URI敲低細胞的細胞毒性作用可以通過抗氧化劑N-乙酰半胱氨酸(NAC)或鐵死亡抑制劑鐵抑素-1(fer1;脂質過氧化清除劑),而細胞凋亡、壞死或自噬抑制劑作用不大(圖1i)。總之,這些結果表明URI與肝癌細胞對TKIs誘導的鐵死亡的耐藥性有關。
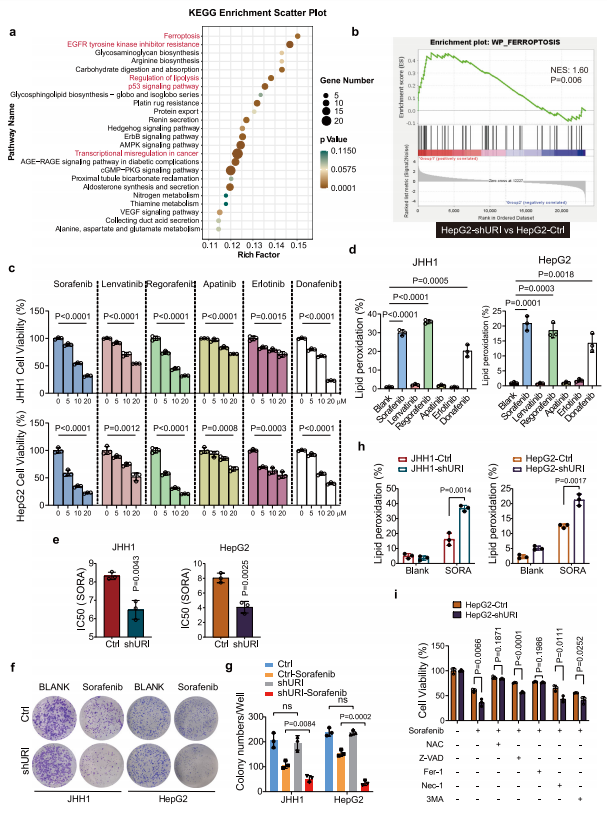
圖1 URI耗竭促進TKIs誘導的癌細胞鐵死亡
2、URI的異位表達重編程了癌細胞中SCD1相關的脂質代謝
癌細胞比正常細胞需要更高水平的脂質代謝,這可以決定細胞對鐵死亡的敏感性。為了檢測URI介導的脂質代謝和脂質組學變化,在HepG2- shURI和HepG2- Ctrl細胞中進行了基于質譜的脂質組學分析(圖2a)。在URI缺失的HepG2細胞中,主要含有單不飽和脂肪酸鏈的最豐富的脂類TGs的相對含量比對照組顯著降低,磷脂酰肌醇(PI(18:1/18:1))和雙甘油酯(DG(16:1/18:1/0:0))的相對含量降低(圖2a, b)。相反,磷脂酰甘油(PG(16:0/16:0))的相對含量在HepG2- shURI細胞中增加(圖2b)。這些結果表明,URI的消耗改變了肝癌細胞的脂質組成。
作者分析HepG2細胞的脂肪酸含量。細胞內細胞脂質過氧化是鐵死亡的一個主要事件,它是由MUFAs和多不飽和脂肪酸(PUFAs)的比例決定的。作者發現,在HepG2- shURI細胞中,SFAs水平比對照組升高,SFAs衍生的代謝產物MUFAs水平,特別是16:1(n-7)和18:1(n-9)MUFAs水平顯著降低(圖2c)。這些結果強烈表明,URI敲除導致從SFAs到MUFAs的約定受損。值得注意的是,作為SCD1活性替代物的16:1(n-7)/16:0和18:1(n-9)/18:0比值在HepG2-shURI細胞中比對照顯著降低(圖2d),表明URI表達與SCD1活性/表達之間存在強烈關聯。
PL-PUFAs易受ROS影響,其脂質過氧化可引發鐵死亡級聯反應。相反,MUFAs可以通過促進PUFAs從血漿膜中位移膜磷脂來抑制這一過程。然后,作者分析HepG2-shURI和HepG2-Ctrl細胞之間磷脂的脂質種類(如PC, PE, PI)。穩態下,HepG2-shURI細胞的磷脂中MUFA比對照組有降低的趨勢(圖2e)。HepG2-shURI細胞中C16:0/C20:4 PL-PUFA含量比對照細胞增加,而HepG2-shURI細胞中PL-MUFA C16:0/C18:1含量降低(圖2f)。因此,雖然HepG2-shURI和對照細胞之間PUFAs沒有明顯變化,但PL-PUFA減少。
從頭合成脂肪酸(FAS)涉及幾種酶的協調作用(圖2g)。接下來,作者測量JHH1和HepG2細胞中FAS通路關鍵酶的mRNA水平,以及它們的URI耗盡酶(圖2h)。與親本細胞相比,JHH1-shURI和HepG2-shURI細胞中參與飽和脂肪酸合成或脂肪酸延伸的酶,包括SCD1、FASN、FADS2、ACACA和ELOVL6的轉錄本顯著減少(圖2h)。總之,這些結果表明URI消耗重編程了肝癌細胞的脂質代謝。
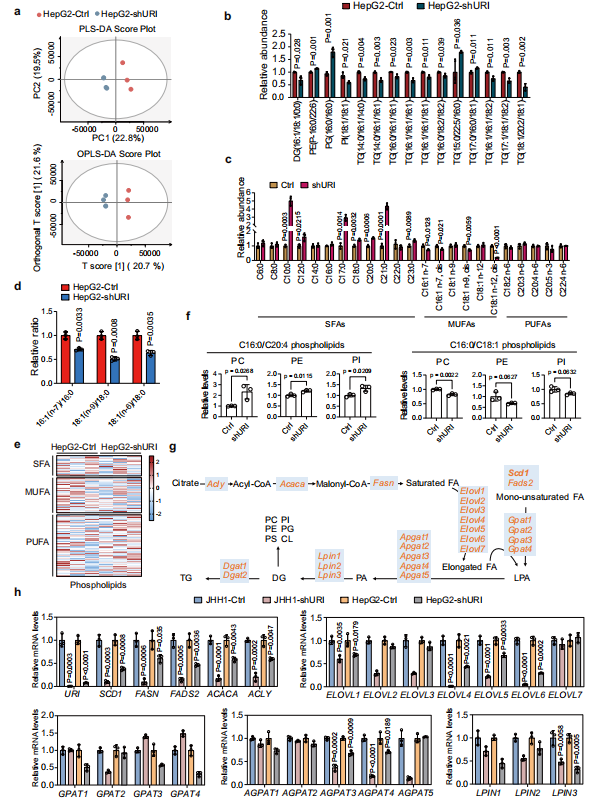
圖2 URI耗竭改變了癌細胞的脂質代謝
3、URI通過SCD1促進對TKIs誘導的鐵死亡的抗性
脂質代謝重編程與癌癥耐藥有關。為了探究URI缺失是否會改變脂質代謝相關酶的蛋白水平,作者分析不同細胞系在基礎狀態下的蛋白水平。雖然在URI敲低的細胞中觀察到ACACA、FASN和FADS2的信使RNA轉錄顯著變化(圖2h),但它們的蛋白水平不受URI的影響(圖3a)。值得注意的是,作者發現shURI細胞中的SCD1蛋白水平低于對照組(圖3a),這與SCD1 mRNA水平的降低是一致的(圖2h),這表明URI可能通過影響SCD1的轉錄來調節其活性。
cystine-import-GSH-GPX4機制是調控鐵死亡的典型途徑。然而,URI缺失在RSL3處理或不處理的JHH1和HepG2細胞中誘導SLC7A11、GCLC、GCLM和GPX4蛋白水平輕度變化(圖3a)。此外,在shURI和對照細胞中,ferrostatin-1可以減少索拉非尼或聯合治療誘導的細胞死亡(圖3b)。與這些結果一致, SCD1抑制劑與索拉非尼聯合在長期克隆實驗中顯示出對腫瘤細胞增殖的協同抑制。抑制鐵死亡可以減輕這種聯合治療誘導的增殖抑制(圖3c, d)。進一步的實驗表明,索拉非尼和SCD1抑制劑聯合使用顯著提高了癌細胞的脂質過氧化,加入ferrostatin-1同樣減少了聯合治療誘導的脂質過氧化(圖3e)。為了證實這些發現,作者將MOCK或Flag-SCD1質粒穩定轉染到癌細胞中。作者的研究結果表明,在URI敲除細胞中,Flag-SCD1的穩定表達足以重新抵抗細胞的鐵死亡(圖3f)。同時,補充外源性SCD1可降低索拉非尼誘導的脂質過氧化(圖3g)。總之,作者的研究結果表明,URI通過上調SCD1來促進對TKIs誘導的鐵凋亡的抗性。
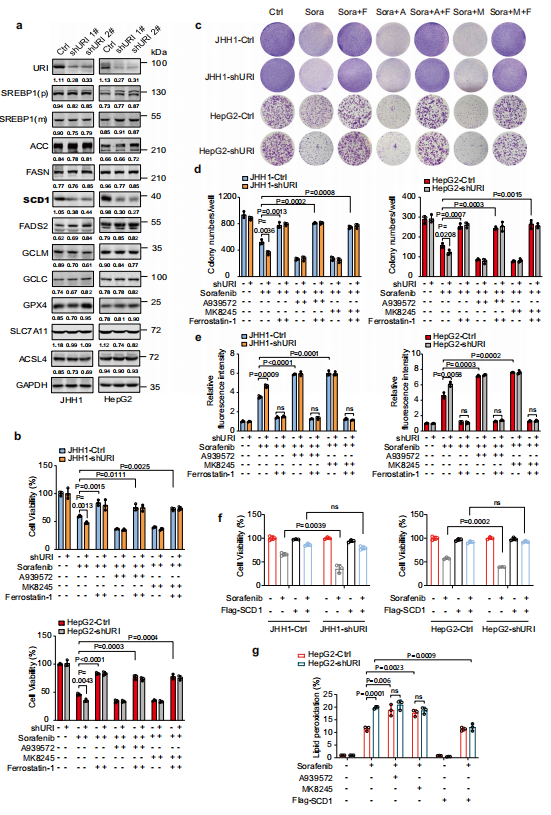
圖3 URI以SCD1依賴的方式減輕TKI誘導的鐵死亡
4、URI在肝癌細胞中通過野生型p53正向調節SCD1轉錄
研究表明,SCD1在轉錄水平上受幾種轉錄因子(TFs)的調控,在翻譯后水平上受蛋白酶體的泛素化和隨后的降解的調控。用His-URI質粒轉染shURI細胞可恢復SCD1蛋白水平(圖4a)。為了研究URI如何上調癌細胞中SCD1的表達,作者首先研究了URI對SCD1蛋白穩定性的影響。這些結果表明,URI上調SCD1表達獨立于泛素蛋白酶體系統。因此,作者假設URI可能通過上調其轉錄來促進SCD1的表達。事實上,報告基因分析顯示,shURI細胞中的SCD1-luc值顯著低于Ctrl細胞(圖4b)。
為了進一步確定哪些轉錄因子介導URI對SCD1的調控,作者將靶向SREBP、ChREBP或LXRα的siRNA轉染到癌細胞中,這是報道的調節SCD1的主要TFs。在干擾這些TFs后,URI介導的SCD1-luc強度差異并未消除(圖4c)。由于ChREBP和LXRα顯示以SREBP依賴的方式增加SCD1的轉錄,作者在HepG2- Ctrl和HepG2- shURI細胞中使用SREBP抗體進行了染色質免疫沉淀(ChIP)分析,發現SREBP在SCD1啟動子區域有多個結合位點(圖4d)。然而,SREBP與SCD1啟動子的結合不受URI表達的影響,與熒光素酶報告基因實驗一致。作者的數據表明這些轉錄因子不參與URI調節SCD1。
先前的研究表明SCD1基因是p53介導的轉錄抑制的靶標。與此一致的是,作者的CUT&Tag分析顯示p53的結合位點存在于SCD1的啟動子區(圖4e)。GSEA發現URI缺失后p53轉錄基因網絡顯著陽性富集(圖4f),RT-PCR進一步證實了這一點(圖4g)。為了驗證p53介導的SCD1表達的作用,作者用多柔比星(一種著名的p53激活劑)處理JHH1和HepG2細胞(表達野生型p53)。結果顯示,Dox處理后SCD1蛋白和SCD1 mRNA表達降低(圖4h, i)。接下來,作者將Mycp53、突變體p53- R273H或p53- R249S轉染到p53-null細胞(Hep3B)中,發現野生型p53顯著下調SCD1蛋白水平。相比之下,DNA接觸突變體(p53-R273H)和構象突變體(p53-R249S)都不影響SCD1蛋白水平(圖4j)。事實上,SCD1基因的5 '側翼區域包含一個與p53結合序列一致的位點(圖4k)。ChIP分析進一步表明,內源性p53結合在SCD1基因的啟動子區域(圖4l)。此外,Dox和nutlin-3都可以激活p53,但不能逆轉p53-敲低狀態下SCD1蛋白水平的升高(圖4m)。作者推測URI上調SCD1表達是由p53介導的。URI缺失增強了p53與SCD1啟動子區域的結合(圖4n)。接下來,作者將p53的siRNA轉染到HepG2-Ctrl和HepG2-shURI細胞中,發現p53的敲低消除了shURI介導的SCD1抑制(圖4n)。作者的數據顯示,URI缺失顯著降低了野生型p53細胞(JHH1和HepG2)中SCD1的表達(圖3a和圖4p)。總之,這些數據表明URI通過抑制野生型p53促進SCD1轉錄水平。
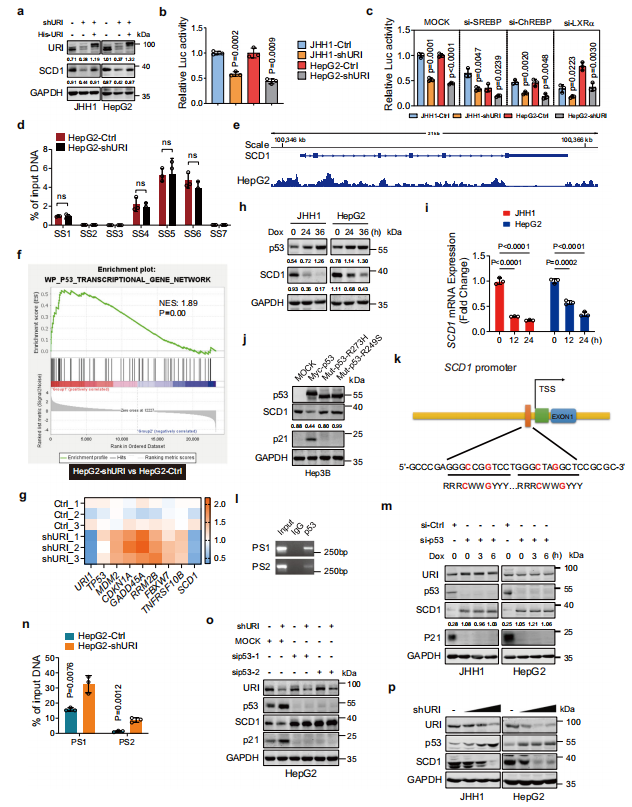
圖4 URI通過抑制p53促進SCD1轉錄
5、URI促進癌細胞中野生型p53的泛素化和降解
接下來,作者將His-URI質粒轉染到JHH1和HepG2細胞中,發現這種處理導致p53蛋白顯著減少,即使在Dox存在的情況下也是如此(圖5a)。為了探究URI介導的p53表達降低的分子機制,作者首先檢測了Ctrl和shURI細胞中p53的mRNA水平。作者的數據顯示,URI缺失不會影響p53的轉錄水平(圖5b)。應激誘導的p53激活主要通過蛋白穩定發生。作者進一步探討URI是否調控p53蛋白的穩定性。事實上,與對照細胞相比,使用環己亞胺(CHX)可以延長JHH1-shURI細胞中p53的半衰期(圖5c)。URI缺失顯著降低了HepG2細胞中野生型p53的泛素化(圖5d)。
由于URI不是E3泛素連接酶,作者假設URI可能會招募E3泛素連接酶來介導p53的泛素化。作者進一步進行了液相色譜串聯質譜(LC-MS/MS)分析,并確定了潛在的URI結合蛋白,包括TRIM28、USP5、USP7和USP14,這些蛋白被報道可以調節p53的泛素化(圖5e)。為了驗證LC-MS/MS的結果,作者通過免疫沉淀法測試了候選蛋白與URI的結合。結果顯示URI和TRIM28之間存在相互作用(圖5f),這與之前的研究結果一致。兩種不同的靶向TRIM28的siRNAs增加了p53蛋白水平,特別是在shURI細胞中(圖5g)。此外,TRIM28的敲低導致MG132處理下p53的泛素化降低(圖5h)。由于TRIM28與MDM2的結合有助于p53蛋白的穩定性,作者探索URI是否會影響TRIM28與MDM2的相互作用。免疫沉淀顯示URI的缺失抑制了TRIM28-MDM2的形成(圖5i)。相反,URI的過表達促進了MDM2和TRIM28的合作(圖5j)。此外,URI缺失降低了TRIM28- MDM2 -p53復合物(圖5i, j),表明URI可能募集E3泛素連接酶TRIM28,促進TRIM28- MDM2對p53蛋白泛素化的作用。體外通過GST-pull down實驗確定URI是否與TRIM28直接相互作用。純化后的GST-URI與His-TRIM28之間的相互作用以濃度依賴性的方式顯著增加(圖5k)。總之,作者的研究結果表明URI與TRIM28相互作用并促進TRIM28- MDM2 -p53復合物的形成,隨后泛素化p53。接下來,作者確定URI/TRIM28對p53蛋白水平的調節是否對鐵死亡有功能影響。TRIM28的敲低顯著促進索拉非尼誘導的細胞死亡和脂質過氧化(圖5l-n)。
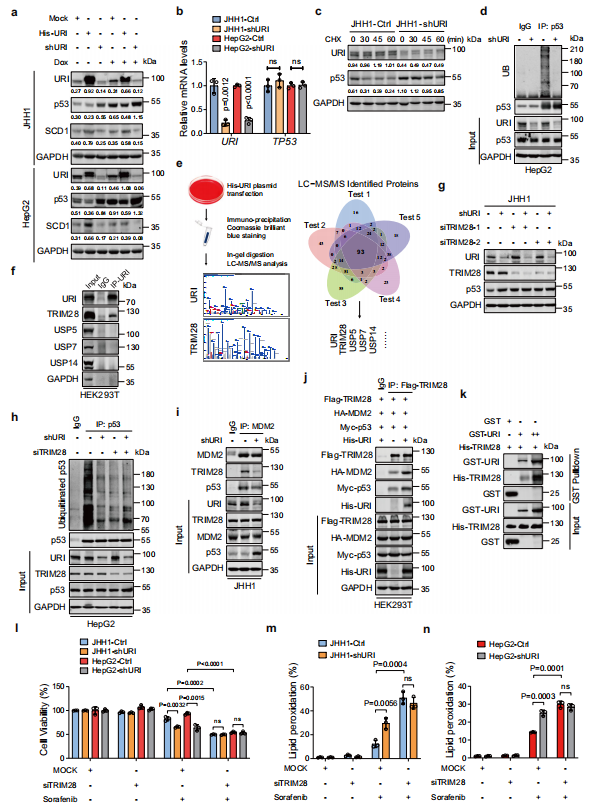
圖5 URI通過與TRIM28相互作用促進p53泛素化和降解
6、SCD1抑制劑與多納非尼在肝癌中的協同作用
作者的上述數據表明,抑制SCD1可顯著逆轉肝癌細胞對TKIs的耐藥性。作者推測TKIs和SCD1抑制劑聯合使用可能對p53野生型癌癥有效。Aramchol是一種新型的SCD1部分抑制劑,在一項2b期臨床試驗中,與其他SCD1抑制劑相比,Aramchol已被證明是非酒精性脂肪性肝炎(NASH)的一種有希望的治療方法,且未導致嚴重不良事件。為了評估aramchol在體內的安全性和抗腫瘤活性,作者使用了嚙齒動物的異種移植模型。結果表明aramchol在小鼠模型中的安全性。
Aramchol在體外對細胞活力沒有明顯的抑制作用(圖6a),與異種移植模型的結果一致。值得注意的是,在長期克隆實驗中,aramchol增強了p53野生型癌細胞對多納非尼的敏感性,多納非尼是一種氘化的索拉非尼衍生物,比索拉非尼更有優勢(圖6b, c)。作者接下來研究了aramchol是否可以在HCC患者衍生的類器官(PDOs)中與多納非尼協同作用。作者也確定了PDOs中p53的狀態(圖6d)。與癌細胞系的結果相似,URI缺失顯著提高了多納非尼在p53野生型PDOs中的敏感性。多納非尼和aramchol聯合使用確實比單獨使用多納非尼表現出更強的細胞毒性作用(圖6e-g)。此外,與單一藥物相比,聯合治療刺激了更高水平的脂質過氧化,這表明aramchol在臨床前模型中促進了多納非尼誘導的鐵死亡(圖6h)。相比之下,p53突變的PDOs對多納非尼的耐藥性更強。
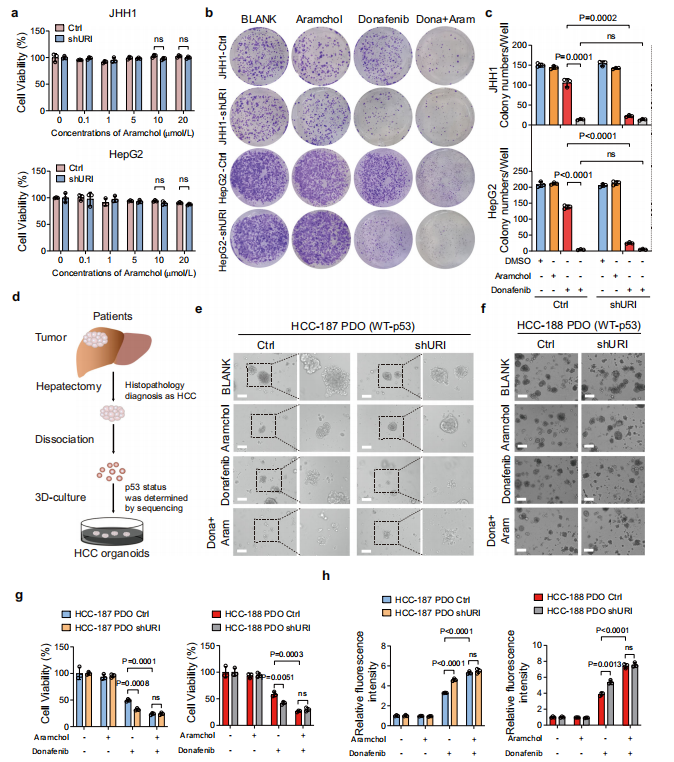
圖6 SCD1抑制劑芳烴可增強多納非尼在癌細胞和患者源性類器官中的療效
作者進一步探討了聯合治療在體內的治療效果(圖7a)。在HepG2-Ctrl和HepG2-shURI細胞異種移植的小鼠中,aramchol和多納非尼聯合使用可完全抑制腫瘤生長,而多納非尼單藥治療HepG2-shURI細胞的異種移植具有有效的治療作用(圖7b-d)。腫瘤切片的免疫組織化學化學分析進一步顯示,aramchol和多納非尼治療小鼠的腫瘤中Ki67降低。此外,研究發現,aramchol和多納非尼聯合使用可促進SCD1和p53的協同喪失(圖7e, f)。綜上所述,這些結果表明SCD1抑制劑aramchol與多納非尼對肝癌具有協同致死性。
考慮到臨床上TKIs主要用于晚期HCC患者。作者延遲給藥,以使HepG2異種移植物的腫瘤生長到適當的大小(圖7g)。再一次,聯合治療引起了明顯的腫瘤控制(圖7h, i)。此外,聯合治療顯著提高了腫瘤中的脂質過氧化水平(圖7j)。因此,多納非尼和aramchol聯合使用可能會獲得顯著的臨床益處,特別是對于野生型p53的HCC患者。
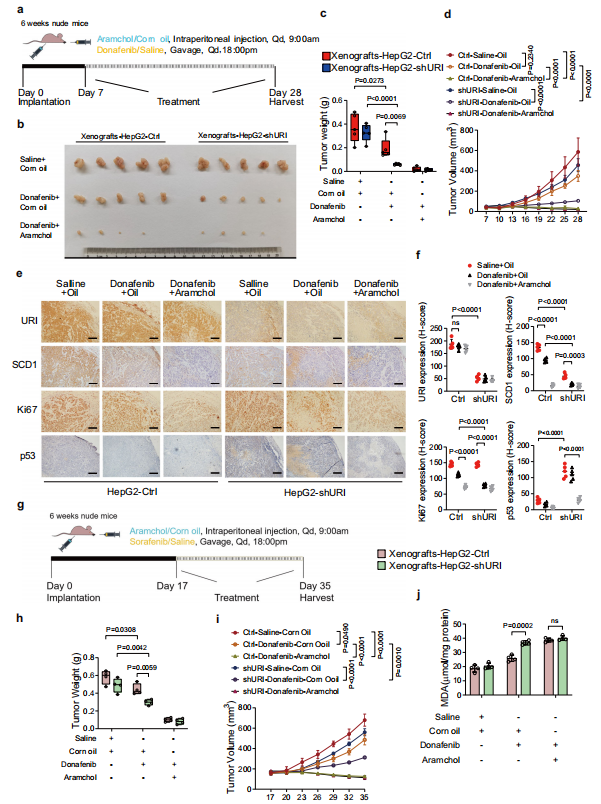
圖7 SCD1抑制劑芳烴可增強多納非尼的體內療效
7、URI和SCD1的高表達與HCC預后惡化和索拉非尼耐藥相關
在作者之前的研究中,作者發現URI在HCC中高表達與腫瘤惡性和患者生存率低相關。為了進一步證實URI-SCD1軸與臨床癌癥治療之間的功能聯系,作者首先利用組織顯微陣列,包括134例晚期HCC樣本(隊列A)。作者在這些樣本中進行多重免疫組織化學/免疫熒光(mIHC/IF)檢測URI和SCD1表達。采用H-score法定量表達,見材料和方法(圖8a)。在隊列A樣本中,URI表達與SCD1表達呈正相關(圖8b, c)。此外,作者根據URI和SCD1的表達將患者分為四組。通過Kaplan-Meier曲線(p < 0.0001, log-rank檢驗)顯示不同亞類間OS的顯著差異(圖8d)。URI和SCD1協同表達較高的患者風險最高,5年OS率為4.3% (HR, 2.65;95% CI, 1.76-3.98)而低URI和SCD1的患者為18.8%。這些結果表明URI和SCD1的聯合表達是HCC預后不良的一個非常有效的預測因子。
由于作者的隊列A中沒有p53狀態數據,為了進一步研究URI-SCD1在不同p53狀態HCC患者中的作用,作者采用了Gao等人招募的一個新的HCC隊列,作者將其命名為“Fudan_HCC_cohort”。通過分析該隊列的WES和轉錄組數據,作者發現在p53-WT狀態的HCC患者中,URIlow腫瘤中的SCD1表達低于URIhigh腫瘤,而其他與鐵死亡相關的分子,如ACSL4、ALOX12、GPX4、SLC7A11和AIFM2沒有明顯改變(圖8e)。此外,p53- WT型HCC患者中URI或SCD1的高表達與較差的臨床預后相關(圖8f, g)。綜上所述,這些結果表明,在野生型p53患者中存在URI-SCD1與HCC患者臨床預后之間的潛在相關性,而在p53突變型HCC患者中則不存在。
為了進一步探討URI-SCD1在索拉非尼耐藥中的作用是否與臨床相關,作者在接受索拉非尼輔助治療的HCC患者隊列(隊列B)中檢測了URI和SCD1的表達。作者的數據顯示URI表達與SCD1表達密切相關,與隊列A的結果一致
然后,作者使用之前的隊列(命名為C隊列),該隊列納入了復發性HCC患者,然后患者接受含有索拉非尼的全身治療。45例患者為p53- WT, 1例患者攜帶p53同義體突變,作者也將其分組為p53- WT。免疫組化法檢測SCD1、URI、p53蛋白表達水平。該隊列中p53-WT患者的總生存率高于p53突變患者(圖8h)。作者發現在p53-WT組中SCD1與URI之間存在顯著相關性,而在p53-突變組中則沒有(圖8i-k)。同時,在接受索拉非尼治療的p53-WT HCC患者中,較高水平的SCD1或URI與預后惡化相關(圖8i - o)。因此,作者的研究結果證明了URI-SCD1軸在p53-WT HCC患者索拉非尼耐藥中的重要作用。
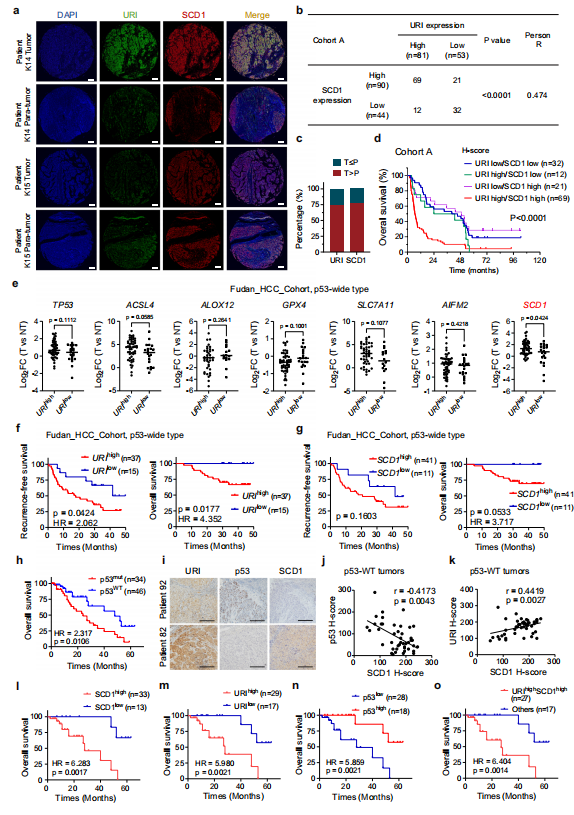
圖8 URI聯合SCD1與晚期HCC患者生存差和索拉非尼耐藥相關
結論
綜上所述,URI以TRIM28-MDM2依賴的方式維持低水平的p53,維持SCD1活性和MUFAs的積累,從而促進癌細胞對TKIs的耐藥性。URI-p53-SCD1軸介導TKIs的耐藥,這可能解釋了為什么p53野生型HCC仍然對TKIs表現出內在耐藥。此外,本研究確定的聯合治療可能代表了約41%具有野生型p53和高水平URI/SCD1的晚期HCC患者的一種有希望的策略(圖9)。本研究為作者后續的臨床試驗提供了理論基礎。
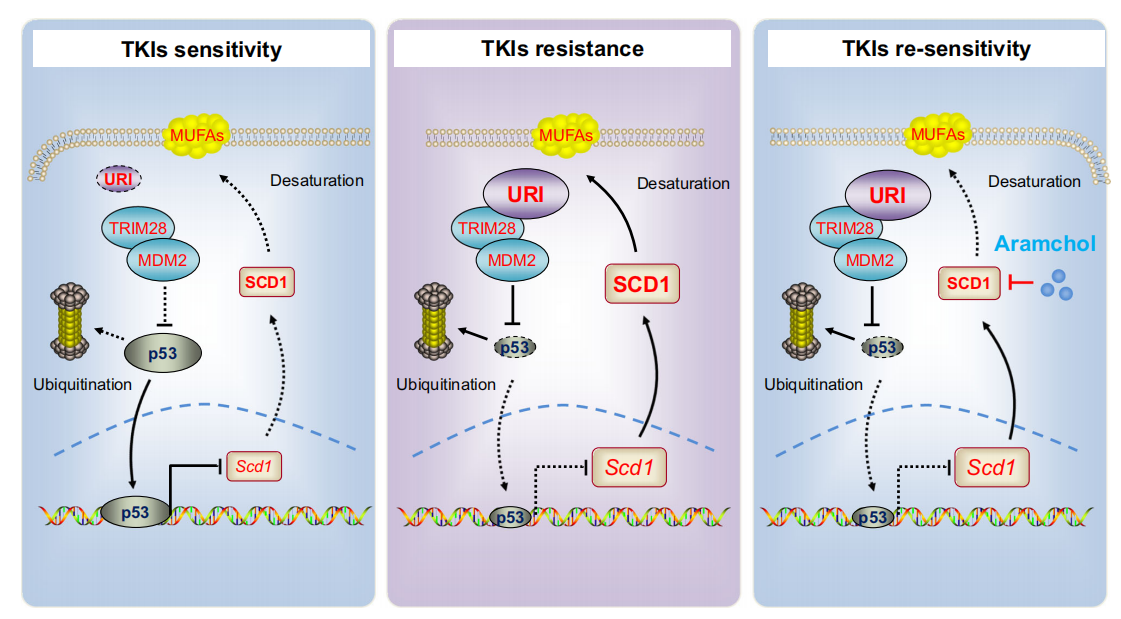
圖9 URI-p53-SCD1軸介導TKIs耐藥和芳烴再敏化肝癌細胞對TKIs的模型
實驗方法
細胞系和細胞培養;人肝細胞癌衍生類器官的培養;實時熒光定量PCR分析;脂質過氧化實驗;細胞增殖、菌落形成和活力測定;流式細胞術;WB、免疫共沉淀和抗體;熒光素酶報告實驗;染色質免疫共沉淀;腫瘤異種移植實驗;免疫組織化學和評分;多路復用免疫組織化學和免疫熒光;RNA測序;CUT&Tag庫建設;LC-MS;靶向中、長鏈脂肪酸定量;蛋白LC-MS/MS分析;DNA電泳;重組Myc-URI和His-TRIM28的生產。
參考文獻
Ding Z, Pan Y, Shang T, Jiang T, Lin Y, Yang C, Pang S, Cui X, Wang Y, Feng XF, Xu M, Pei M, Chen Y, Li X, Ding J, Tan Y, Wang H, Dong L, Wang L. URI alleviates tyrosine kinase inhibitors-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers. Nat Commun. 2023 Oct 7;14(1):6269. doi: 10.1038/s41467-023-41852-z. PMID: 37805657; PMCID: PMC10560259.