






靶向m6A閱讀器YTHDF1增強結直腸癌的抗腫瘤免疫和抗PD-1療效
摘要
N6-甲基腺苷(m6A)在腫瘤免疫微環境(TIME)中的作用仍有待進一步研究。在這里,作者闡明YTHN6-甲基腺苷RNA結合蛋白1(YTHDF1)在大腸癌(CRC)TIME中的功能和機制。在組織微陣列(N=408)和TCGA(N=526)隊列中評估YTHDF1的臨床意義。在同基因腫瘤、腸特異性Ythdf1敲入小鼠和人源化小鼠中測定YTHDF1功能。采用單細胞RNA-seq(scRNA-seq)分析TIME。采用甲基化RNA免疫沉淀法測序(MeRIP-seq)、RNA測序(RNA-seq)和核糖體測序(Ribo-seq)鑒定YTHDF1直接靶標。用囊泡狀納米顆粒(VNPs)包封YTHDF1-siRNA進行YTHDF1體內沉默。在TCGA-CRC中,YTHDF1表達與干擾素-γ基因特征負相關。與此同時,在獨立的組織微陣列隊列中,YTHDF1蛋白與CD8+ T細胞浸潤呈負相關,暗示其在TIME中的作用。基因缺失Ythdf1增強CT26(MSS-CRC)和MC38(MSI-HCRC)同基因腫瘤的抗腫瘤免疫,而在偶氮甲烷-葡聚糖-硫酸鈉或ApcMin/+模型中,Ythdf1敲入蛋白促進免疫抑制TIME促進結直腸癌。scRNA-seq發現,在Ythdf1基因敲除腫瘤中,髓源性抑制細胞(MDSCs)減少,同時細胞毒性T細胞增加。綜合MeRIP-seq、RNA-seq和核糖核酸-seq發現p65/Rela是YTHDF1的靶標。YTHDF1促進p65翻譯上調CXCL1,從而增加MDSC通過CXCL1-CXCR2軸的遷移。增加的MSDCs反過來在時間上拮抗功能性CD8+ T細胞。重要的是,通過CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)或VNPs-siYTHDF1靶向YTHDF1可提高MSI-HCRC的抗PD1療效,并克服MSSCRC的抗PD1耐藥性。YTHDF1通過M6A-P65-CXCL1/CXCR2軸損害抗腫瘤免疫,促進結直腸癌,并在免疫檢查點阻斷治療中作為治療靶點。本文于2023年8月發表在《Gut》IF:24.5期刊上。
技術路線

主要實驗結果
1、YTHDF1與結直腸癌中IFN-γ相關基因特征降低和預后不良相關
通過使用TCGA數據分析21個已知的m6A調控子的拷貝數變化,作者鑒定出YTHDF1是m6A調控子在近80%的CRC腫瘤中顯示拷貝數增加或擴增中最高。這種拷貝數的增加也與mRNA表達和蛋白表達的上調相一致,提示YTHDF1具有促進CRC的功能。為建立抗腫瘤免疫與m6A調節因子之間的聯系,該研究隨后使用GSEA分析m6A調節因子與IFN-γ反應基因標記之間的相關性。作者發現YTHDF1與IFN-γ反應通路呈顯著負相關(q<0.0001)。值得注意的是,IFN-γ反應與抗腫瘤免疫的誘導和對ICB治療的反應有關。一致地,YTHDF1的表達與18 IFN-γ相關的基因標記呈強反相關(圖1A),這預測多種癌癥類型的抗PD1反應性。此外,CD8A或CD8+ T細胞特征的表達與YTHDF1表達呈負相關。與TCGA數據一致,來自CRC TMA隊列的TMA IHC染色顯示,隊列I(p<0.001,r= -0.248,n=206)(圖1B)和隊列II(p<0.0001,r= -0.269,n=202)中,YTHDF1蛋白的高表達與CD8+ T細胞的低浸潤相關(圖1C)。這些數據強烈暗示,m6A閱讀器YTHDF1與抗腫瘤免疫功能受損和ICB治療效果降低有關。
與CD8+ T細胞浸潤低與預后不良相關的觀點一致,YTHDF1高表達(54.3%,100/184)預測CRC患者生存不良(p<0.01,log-rank檢驗)。多因素Cox回歸分析證實,YTHDF1是隊列II中結直腸癌的獨立預后因素(HR,1.764;95%可信區間1.058-2.939;p<0.05)。在隊列I中,通過多變量Cox回歸分析進一步驗證這些發現。
2、單細胞轉錄組學揭示YTHDF1誘導的免疫抑制
為研究YTHDF1在調節抗腫瘤免疫中的作用,作者使用CRISPR-Cas9系統敲除MC38小鼠MSI-H CRC細胞中的Ythdf1(Ythdf1-KO),并將細胞注射到同基因C57BL6小鼠(圖1D)。發現與對照組(NC)相比,敲除Ythdf1后,腫瘤體積和重量都減少(圖1E)。為了解Ythdf1-KO是否影響TIME,作者從腫瘤中分離出CD45+免疫細胞,并進行單細胞RNA-seq(scRNA-seq)(NC:1480個細胞;Ythdf1-KO:1816個細胞)。與NC組相比,含有Ythdf1-KO的腫瘤表現出粒細胞髓源性抑制細胞(G-MDSCs,簇3)和中性粒細胞的顯著減少(圖1F)。相比之下,Ythdf1-KO腫瘤中T細胞和NK細胞大量增加(圖1F)。作者進一步將T細胞和NK細胞重新聚集為CD4+ T、CD8+ T、NKT和NK細胞亞群,并發現與對照組相比,它們在Ythdf1-KO腫瘤中同時增加(圖1F)。因此,作者推測YTHDF1可以通過誘導MDSC積累來抑制抗腫瘤免疫。通過檢測MDSCs的功能標記物Il1b、Arg2、Cxcr2和Ccr2,這些基因主要富集于MDSC簇(簇3和簇4),特別是來自未敲除Ythdf1的腫瘤(圖1G)。因此,來自該同基因模型的數據支持YTHDF1在結直腸癌中的免疫抑制功能。

圖1 在免疫功能正常的小鼠中通過scRNA-seq驗證YTH N6-甲基腺苷RNA結合蛋白1(YTHDF1)與結直腸癌(CRC)患者的免疫抑制微環境相關
3、敲除Ythdf1可減少MDSCs,但增加細胞毒性T細胞浸潤
為驗證在scRNA-seq分析中的發現,作者用流式細胞術測定MC38同基因小鼠腫瘤浸潤免疫細胞的組成,證實敲除Ythdf1顯著抑制腫瘤的重量和體積(圖1C)。并且流式細胞術顯示,敲除Ythdf1減少MDSCs,但增加腫瘤中的CD8+ T和CD4+ T細胞(圖1H,I)。在MDSCs中,G-MDSCs是主要的亞群,敲除Ythdf1導致G-MDSCs顯著減少(圖1I)。與MDSCs的免疫抑制功能一致,作者觀察到Ythdf1-KO組功能T細胞顯著增加,包括IFN-γ+ CD8+ T細胞、顆粒酶B+ CD8+ T細胞和IFN-γ+ CD4+ T細胞(圖1H,J)。
接下來,作者在CT26(MSS-CRC)中與Ythdf1-KO進行實驗,以驗證YTHDF1在調節抗腫瘤免疫中的作用。正如預期的那樣,Ythdf1-KO導致CT26同基因小鼠腫瘤體積和重量減小(圖2A,B),同時功能T細胞減少和MDSCs積累(圖2C,D)。免疫熒光染色證實,Ythdf1基因敲除后,MC38和CT26同基因腫瘤中MDSCs(CD11b+ Gr-1+)的浸潤減少(圖2E)。總的來說,結直腸癌細胞中Ythdf1的缺失減少MDSCs,增加功能性T細胞的浸潤。這些發現與臨床數據一致,顯示YTHDF1與CD8+ T細胞和IFN-γ相關的特征(圖1A-C)。
作者想知道在Ythdf1-KO中減弱的腫瘤形成是否依賴于CD8+ T細胞抗腫瘤免疫。為解決這個問題,作者在MC38同基因模型中使用抗CD8抗體去除CD8+ T細胞。與假設一致,CD8+ T細胞的消耗恢復Ythdf1-KO腫瘤的生長(圖2F,G),表明Ythdf1-KO的腫瘤抑制功能至少部分依賴于CD8+ T細胞。這在CT26同基因小鼠中得到證實,表明抗CD8抗體治療可以挽救Ythdf1-KO組中被抑制的腫瘤生長(圖2H,I)。總之,Ythdf1敲除通過誘導CD8+ T細胞依賴性抗腫瘤免疫抑制結直腸癌的生長。

圖2 YTH N6-甲基腺苷RNA結合蛋白1(Ythdf1)敲除通過減少髓源性抑制細胞(MDSC)和增加同源腫瘤中的功能性T細胞來誘導抗腫瘤免疫,這種作用被CD8+ T細胞消耗逆轉
4、腸道特異性Ythdf1敲入促進小鼠結直腸腫瘤發生并抑制抗腫瘤免疫
為驗證YTHDF1在自發性結直腸腫瘤發生中的作用,作者構建腸道特異性的Ythdf1敲入小鼠(Ythdf1loxp/loxp CDX2-cre),并通過AOM/DSS治療在這些小鼠中啟動CRC(圖3A),發現在AOM/DSS模型中,Ythdf1的過表達導致結腸腫瘤數量和大小增加(圖3C)。流式細胞術顯示,與野生型小鼠相比,Ythdf1敲入小鼠結腸腫瘤中MDSC浸潤增加,NK、CD4+ T和CD8+ T細胞減少(圖3D)。此外,作者發現,通過檢測顆粒酶B、INF-γ和TNF-α的表達,Ythdf1敲入降低功能性T細胞的比例(圖3E)。
接下來,作者通過建立ApcMin/+ Ythdf1loxp/loxp CDX2-cre小鼠,試圖在ApcMin/+驅動的自發性CRC中驗證這些結果(圖3B)。與此一致的是,腸道特異性敲入Ythdf1的ApcMin/+小鼠的結腸腫瘤明顯大于野生型小鼠(圖3C)。腫瘤浸潤性免疫細胞分析發現ApcMin/+敲入Ythdf1后,腫瘤中NK、CD4+ T和CD8+ T細胞的浸潤顯著減少,同時MDSCs誘導增加(圖3F)。此外,Ythdf1敲入降低ApcMin/+小鼠的顆粒酶B+、INF-γ+或TNF-α+ T細胞(圖3G)。總之,這些結果支持YTHDF1培養促進自發性CRC的免疫抑制微環境。
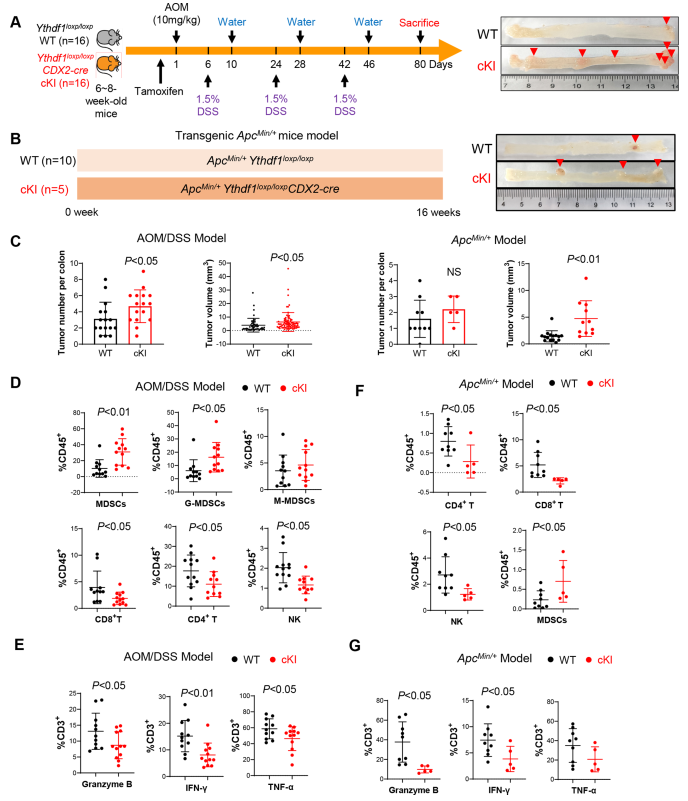
圖3 腸道特異性YTH N6-甲基腺苷RNA結合蛋白1(Ythdf1)敲入促進小鼠結腸腫瘤發生并抑制抗腫瘤免疫。
5、通過VNP-siYTHDF1靶向YTHDF1增強CD34+人源化小鼠的抗腫瘤免疫
為證實YTHDF1在調節人抗腫瘤免疫應答中的作用,作者建立CD34+人源化小鼠模型小鼠外周血單個核細胞(PBMCs)含有>20%的人CD45+細胞(圖4A)。為在體內靶向YTHDF1,作者開發VNPs來攜帶針對YTHDF1的siRNA。在腫瘤達到50-100 mm3后,攜帶人結直腸癌HCT116異種移植物的人源化NSG小鼠接受VNP-siNC或-siYTHDF1治療(圖4B)。與VNP-siNC相比,VNP-siYTHDF1顯著抑制腫瘤體積和重量(圖4B,C)。進行流式細胞術分析TIME(圖4D)。VNP-siYTHDF1減少MDSC浸潤,但增加CD4+ T細胞、CD8+ T細胞和NK細胞積聚(圖4E)。此外,在接受VNP-siYTHDF1的腫瘤中發現更多的IFN-γ+、TNF-α+和顆粒酶B+ CD8+ T細胞(圖4E)。因此,利用VNP-siYTHDF1靶向YTHDF1是增強人源化小鼠抗腫瘤免疫的一種安全有效的手段。

圖4 VNP-siYTHDF1增強CD34+人源化小鼠的抗腫瘤免疫
6、YTHDF1促進p65翻譯激活TNF/NF-κB信號傳導
為確定YTHDF1引發免疫抑制的分子機制,作者在敲除或不敲除YTHDF1的CRC細胞中進行Ribo-seq。Ribo-seq數據顯示,YTHDF1的缺失與TNF信號的失活顯著相關(圖5A)。因此,YTHDF1敲除減少參與TNF信號傳導的核糖體保護片段基因豐度(圖5A)。因此,YTHDF1可以通過促進蛋白翻譯調節TNF/NF-κB信號傳導。由于YTHDF1具有m6A閱讀器的功能,作者接下來進行m6A免疫沉淀測序(MeRIP-seq)以確定m6A修飾的轉錄本。通過篩選參與TNF信號傳導的mRNA中的m6A峰,作者確定兩個靠近p65 mRNA終止密碼子的m6A位點(圖5B),并通過MeRIP-qPCR驗證這一點(圖5C)。重要的是,通過RNA免疫沉淀(RIP)測序和抗YTHDF1抗體的RIP-qPCR發現YTHDF1和p65 mRNA之間的直接相互作用(圖5D)。因此,p65 mRNA是YTHDF1的直接靶點。作者發現在CT26和MC38細胞中,敲除Ythdf1降低p65蛋白的表達,尤其是核p65的表達,而不影響NF-κB通路的其他調節因子如IKKα和iκBα的表達(圖5E)。在人類結直腸癌細胞中獲得一致的結果,顯示野生型YTHDF1過表達,而非功能失調突變體,p65蛋白表達升高;相反,YTHDF1敲低會減弱人類結直腸癌細胞中的p65蛋白(圖5F)。使用抗YTHDF1抗體的RIP-qPCR也證實人結直腸癌細胞中YTHDF1與p65 mRNA之間的直接相互作用(圖5G)。接下來,作者試圖在體內驗證YTHDF1和p65的關聯。在敲入Ythdf1的小鼠中,結腸腫瘤中的p65和phospho-p65蛋白均升高(圖5H)。總之,YTHDF1促進p65蛋白的表達,在體外和體內激活TNF和NF-κB信號。
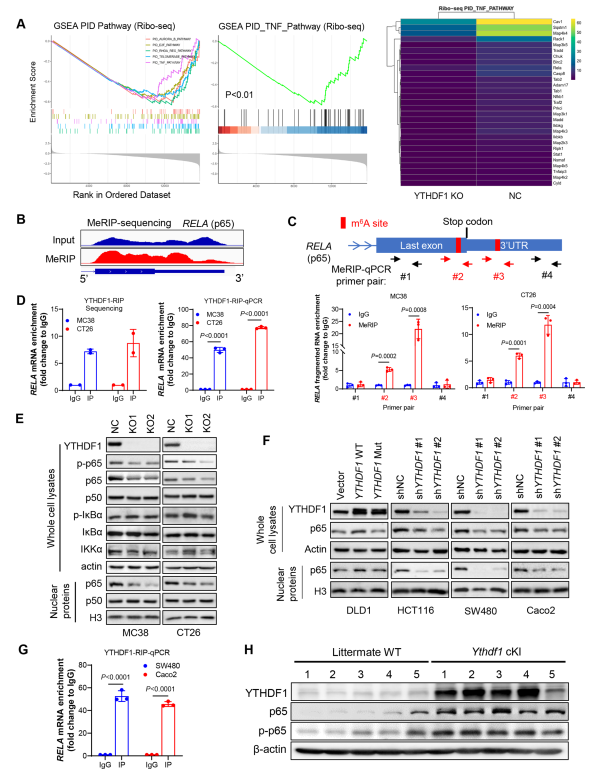
圖5 YTH N6-甲基腺苷RNA結合蛋白1(YTHDF1)通過促進RELA (p65) mRNA翻譯促進CRC中TNF/NF-kB信號傳導
7、YTHDF1通過p65-CXCL1軸促進MDSC遷移
為了解YTHDF1誘導的p65與其免疫抑制之間的聯系,作者在CRC細胞、腫瘤裂解物和同源腫瘤小鼠血清的條件培養基中進行細胞因子多重免疫測定,檢測23種不同的小鼠細胞因子。在這些細胞因子中,Ythdf1-KO持續降低CXCL1(圖6A),并且通過ELISA檢測證實CXCL1的這種降低(圖6B)。據報道,CXCL1是NF-κB信號傳導的轉錄靶點,它通過與其受體CXCR2的相互作用促進MDSC趨化。鑒于Ythdf1-KO在CRC TIME減少MDSC的浸潤,作者想知道YTHDF1是否調節MDSC的遷移。因此,作者進行MDSC體外遷移實驗,發現來自野生型CRC細胞的條件培養基增強MDSC的遷移,而這種遷移被Ythdf1的敲除所破壞(圖6C)。通過CXCR2抑制劑SB265610阻斷CXCL1-CXCR2相互作用,消除對照組和Ythdf1-KO培養上清在介導MDSC遷移方面的差異(圖6C)。因此,YTHDF1通過CXCL1/CXCR2軸促進MDSC遷移。接下來,作者想知道YTHDF1是否可以調節Cxcl1 mRNA的表達。正如預期的那樣,敲除Ythdf1顯著抑制小鼠(圖6D)和人CRC細胞系中Cxcl1 mRNA的水平。因此,研究結果支持YTHDF1-p65-CXCL1/CXCR2軸介導CRC中MDSC的遷移。
接下來,作者研究YTHDF1是否影響MDSC功能。從MC38同基因腫瘤中分離CD11b+ Gr-1+ MDSCs,然后與T細胞體外共培養(圖6E)。從對照腫瘤中分離的MDSCs抑制T細胞增殖;然而,與來自對照組的MDSCs相比,來自Ythdf1-KO腫瘤的MDSCs對CD8+ T細胞和CD4+ T細胞的增殖抑制活性明顯降低(圖6F,G)。這些數據驗證MDSCs對關鍵效應細胞(包括CD8+ T細胞和CD4+ T細胞)的免疫抑制功能,并支持表達YTHDF1的CRC招募功能性MDSCs。
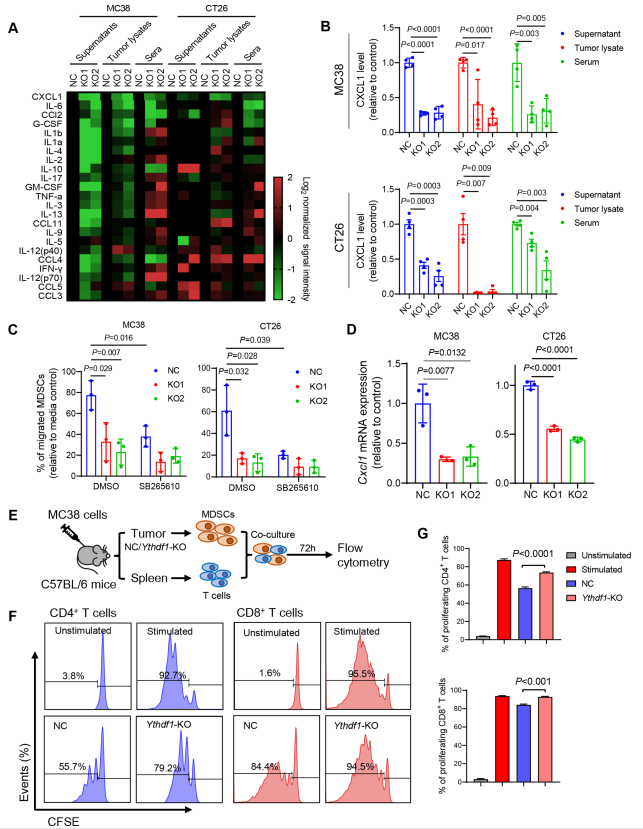
圖6 YTH N6-甲基腺苷RNA結合蛋白1(Ythdf1)的缺失通過減少CXCL1的分泌來促進髓源性抑制細胞(MDSCs)的減少。
8、YTHDF1是CRC免疫治療的潛在治療靶點
考慮到MDSCs浸潤減少已被報道與各種癌癥類型的免疫治療效果增強相關,作者試圖測試靶向YTHDF1是否能增強CRC的抗PD1治療。正如預期的那樣,作者發現敲除Ythdf1增強MC38 (MSI-H)同基因腫瘤中抗PD1的功效,延長荷瘤小鼠的生存期(圖7A)。作者進一步利用VNPs系統將特異性Ythdf1-siRNA傳遞到腫瘤中。當MC38同基因腫瘤達到50~100 mm3時,作者用VNP-siYthdf1(或VNP-siNC)和抗PD1(或IgG)治療小鼠。與VNP-siNC相比,VNP-siYthdf1顯著抑制MC38腫瘤生長(圖7B,C)。值得注意的是,VNP-siYthdf1與anti-PD1聯合使用對腫瘤生長的抑制作用最強(圖7B,C)。
基于同基因CT26(MSS CRC)腫瘤模型,作者進一步研究靶向YTHDF1是否可以克服MSS CRC的抗PD1耐藥性。因此,將敲除Ythdf1的CT26細胞注射到同基因小鼠體內,并用PD1抗體處理,發現敲除Ythdf1顯著增強CT26同基因腫瘤的抗PD1治療效果,否則這些腫瘤對ICB治療無反應(圖7D,E)。
流式細胞術分析進一步顯示,在CT26和MC38同基因模型中,Ythdf1沉默和抗PD1聯合使用可顯著增加腫瘤浸潤的功能性CD8+ T細胞,包括IFN-γ+ CD8+ T細胞和顆粒酶B+ CD8+ T細胞(圖7F,G)。此外,聯合治療顯著減少MDSCs的積累,同時誘導CD4+ T細胞和CD8+ T細胞(圖7G)。因此,靶向YTHDF1不僅可以增強ICB在MSI-H型CRC中的治療效果,還可以通過抑制MDSCs的募集和改善CD8+ T細胞的功能來克服MSS型CRC中的ICB耐藥性。
在這里,作者發現在癌細胞中消耗Ythdf1會抑制腫瘤內的MDSCs,并減輕MDSCs對效應細胞的免疫抑制功能。MDSCs和T細胞共培養研究表明,與對照腫瘤相比,來自Ythdf1-KO腫瘤的MDSCs抑制CD8+ T細胞和CD4+ T細胞增殖的能力受損。因此,研究結果支持YTHDF1介導功能性MSDCs的募集,這抑制CRC中效應T細胞的功能和增殖,導致免疫監視功能受損(圖7H)。

圖7 靶向YTH N6-甲基腺苷RNA結合蛋白1(YTHDF1)增強抗PD1阻斷治療在微衛星不穩定性高(MSI-H)和微衛星穩定型結直腸癌(CRC)中的應用
綜上所述,YTHDF1在結直腸癌中的表達通過激活m6 A-p65-CXCL1軸募集免疫抑制性MDSCs抑制T細胞,從而促進結直腸癌的發生。靶向YTHDF1加抗PD1治療對結直腸癌有良好的抗腫瘤療效,證實YTHDF1是結直腸癌的潛在治療靶點。
實驗方法
人源化小鼠模型,TCGA數據分析,CRISPR/Cas9敲除,穩定細胞系過表達,同基因小鼠模型,RT-qPCR,免疫組織化學,免疫熒光,Western blot,酶聯免疫測定(ELISA),RNA干擾,囊泡樣plga基納米顆粒(VNP)配方,流式細胞術分析,單細胞RNA測序,MDSC分離及遷移試驗,細胞因子多重免疫測定,核糖體測序,MeRIP測序和MeRIP-qPCR,RIP-測序或RIP-qPCR,肝腎功能指標測定
參考文獻
Bao Y, Zhai J, Chen H, et al. Targeting m6A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut. 2023; 72 (8): 1497-1509. doi: 10.1136/gutjnl-2022-328845