






一把鐵做的雙刃劍——在壓力應激中的隨機應變
鐵是造血和各種關鍵細胞功能所必需的礦物質。在許多疾病和失調癥中,人們越來越多地觀察到鐵代謝的改變,但對鐵代謝受損對細胞的影響仍缺乏全面的機理認識。本文中,作者研究鐵過載或缺鐵對細胞應激反應和自噬的影響,它們可調控細胞穩態和存活。研究結果顯示急性鐵負荷導致線粒體ROS(mtROS)的產生和損傷增加、脂質過氧化、自噬通量受損和鐵死亡。鐵誘導的mROS過度產生是脂質過氧化增加、自噬功能受損和誘導鐵死亡的機制。鐵過量誘導的鐵死亡依賴于細胞類型,并受激活轉錄因子4(ATF4)的調控。上調ATF4可減輕鐵誘導的自噬功能障礙和鐵死亡,而沉默ATF4表達則會損害自噬功能,導致mtROS生成增加和鐵死亡。利用自噬缺陷的肝細胞和不同的自噬抑制劑,發現自噬損傷使細胞對鐵誘導的鐵死亡更敏感。相反,缺鐵會激活內質網(ER)應激反應,減少自噬并誘導細胞凋亡。缺鐵相關的自噬減少是由于ER應激,表現為使用4-PBA降低ER應激反應可改善自噬通量。缺鐵導致自噬減少的機制是由于溶酶體膜蛋白翻譯后成熟受損導致溶酶體生物發生紊亂。總之,鐵過量和缺鐵失會導致不同形式的細胞應激和死亡,部分是通過自噬功能受損這一共同機制造成的。本文于2022年7月發表在《Redox Biology》IF:11.8期刊上。
技術路線

主要實驗結果:
1、過量的鐵誘導氧化反應和自噬的變化但不影響ER應激蛋白
由于細胞對鐵含量改變的反應不同,作者確定了鐵過量對多種細胞類型的影響,包括人肝癌細胞系HepG2、非轉化的小鼠肝細胞系AML12和小鼠巨噬細胞系RAW264.7,以及原代細胞P-Hepa和人主動脈內皮細胞HAECs(圖1)。之所以選擇這些細胞類型,是因為肝細胞和巨噬細胞在鐵的儲存和再循環中起著關鍵作用,血管內皮細胞是鐵超載介導損傷的主要靶點之一。
所有的細胞都顯示NRF2表達升高以響應FAC鐵過載誘導,與HepG2和RAW264.7細胞相比,AML12細胞對NRF2的誘導更為急性和強烈(圖1A)。ATF4在AML12和RAW264.7細胞中的誘導作用明顯大于HepG2細胞(圖1A)。HepG2和AML12細胞中沒有誘導包括CHOP、Grp78和P-PERK在內的ER應激標志物,只有RAW264.7細胞中CHOP表達增加,總體上表明過量鐵不誘導ER應激(圖1A)。HepG2細胞中LC3B-II、活性亞型和p62的表達增加,相反,在AML12和RAW264.7細胞中LC3B的表達無明顯改變(圖1A)。盡管在AML12細胞中,FTH1在較早的時間點被強烈誘導,但細胞環境依賴性的自噬調節并不是由于基于FTH1誘導的鐵負荷的大小(圖1A)。在原代培養細胞HAECs和P-Hepa中進一步檢測過量鐵對NRF2、ATF4和自噬因子表達的影響。兩種細胞類型NRF2、p62和LC3B-II的表達均增加,但ATF4的表達未增加(圖1B)。與細胞系相比,鐵負荷對原代細胞中自噬標志物LC3B-II和p62的影響更強。通過使用FeDex處理WT小鼠,在肝臟(過量鐵沉積的主要部位)中測定了急性鐵負荷對氧化和自噬標志物表達的體內影響。FeDex注射顯著增加FTH1的表達,同樣,NRF2、LC3B和p62的表達顯著增加,但ATF4的表達沒有變化(圖1C)。總之,結果表明,急性鐵超載會誘導氧化反應和自噬蛋白表達的改變,但不會引起ER應激反應。

圖1 過量的鐵誘導應激和自噬蛋白的差異表達
2、鐵過量引起自噬通量的損害,從而增強鐵凋亡
LC3B-II表達升高可提示自噬刺激或自噬通量受損。通過使用自噬抑制劑BafA1或Chlq,確定P-Hepa、HepG2細胞和HAECs中LC3B-II表達的增加是否是自噬通量降低的結果。FAC誘導P-Hepa中p62和LC3B-II的表達在8 h和24 h時間點均顯著增加;與BafA1單獨處理相比,FAC/BafA1共處理并未進一步增加p62或LC3B-II水平(圖2A)。FAC處理誘導P-Hepa中LC3B陽性點狀細胞大量聚集;一些鐵處理的P-Hepa顯示強而彌漫性的LC3B染色(箭頭)貫穿整個細胞質以及細胞核的收縮;高倍鏡下,這些細胞含有大空泡,完整性完全喪失(圖2B),表明細胞死亡。重要的是,鐵誘導的LC3B-II積累可被Fer-1阻斷(圖2C)。這些結果表明,過量的鐵誘導脂質過氧化依賴性鐵死亡,而清除脂質過氧化物可挽救鐵誘導的自噬流抑制。

圖2鐵過量誘導脂質過氧化依賴性自噬損傷
鑒于過量的鐵只在自噬功能障礙的細胞中誘導鐵死亡,而自噬在清除氧化分子和受損細胞器方面起著關鍵作用,所有作者利用P-Hepa 細胞模型想知道自噬功能受損是否是鐵過量誘導鐵死亡的原因。與單獨使用FAC相比,FAC/Chlq共處理導致PI+細胞的百分比增加約3.7倍(圖3A)。同樣,與單獨使用FAC相比,FAC和自噬抑制劑E64d/epstatin A共處理導致PI陽性細胞的百分比增加2.6倍(圖3B)。沒有利用BafA1來確定鐵誘導的細胞死亡,因為它通過提高核內體和溶酶體的pH值來干擾細胞內鐵的活動。用4-OHT處理P-Hepa敲除Atg5(ATG5Δhep)(圖3C)。與上述自噬通量受損的情況相一致,自噬缺陷也增強FAC誘導的鐵死亡,表現為ATG5Δhep細胞中PI陽性細胞的死亡率高于對照細胞(圖3D)。這些發現表明,至少在鐵過量的情況下,自噬功能障礙會增強鐵死亡。
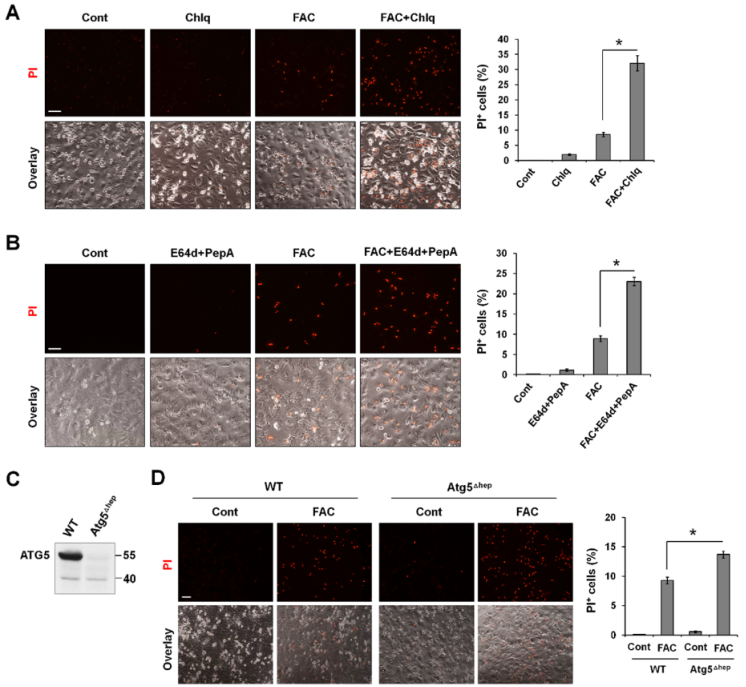
圖3 受損的自噬增強鐵誘導的鐵死亡
3、鐵誘導的mtROS產生是脂質過氧化依賴性鐵死亡的基礎
鐵催化的ROS破壞細胞器。TEM成像顯示,經FAC處理的P-Hepa細胞的線粒體破裂,嵴(箭頭)缺失,這是鐵死亡的一個重要特征(圖4A)。作者推測鐵在線粒體中的負載催化了介導線粒體損傷的mtROS的產生。為探討這種可能性,用Mito-FerroGreen染色Fe2+,并用MitoSOX檢測mtROS。在FAC處理6小時后,觀察到線粒體Fe2+和ROS的強烈疊加(圖4B)。通過使用熒光探針JC-1檢測發現FAC處理導致MMP減少,紅色熒光強度明顯降低(圖4C)。這些發現表明,線粒體中的急性鐵負荷會導致mtROS生成過多和MMP 損失,從而導致線粒體損傷。
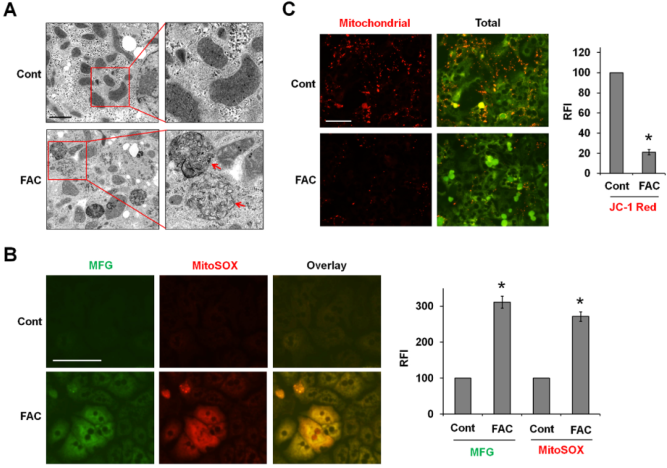
圖4鐵離子被裝載在線粒體中,誘導ROS的產生、MMP的丟失和線粒體損傷
上述研究結果表明,過量產生的線粒體ROS可能是FAC誘導鐵死亡的原因。作者研究了常用的線粒體靶向ROS清除劑MitoTEMPO(MT)是否能挽救鐵誘導的鐵死亡。重要的是,在P-Hepa中,MT和Fer-1都能消除FAC誘導的鐵死亡(圖5A)。熒光探針BODIPY 581/591在氧化時會變成綠色,通過對其染色,證明MT阻斷鐵介導的脂質過氧化的效果與鐵氧化抑制劑Fer-1不相上下(圖5B)。MT還能阻止鐵誘導的LC3B-II和p62的積累(圖5C),這表明自噬通量得到了改善。這些數據表明,mtROS的過度產生增加了脂質過氧化,這是鐵誘導鐵變態反應的基本機制。
魚藤酮(Rotenone)是線粒體復合物I的選擇性抑制劑,在高濃度下可誘導mtROS生成并導致細胞凋亡。作者探究低劑量的魚藤酮與無毒的FAC濃度相結合誘導的mtROS升高是否會使肝細胞對鐵依賴性鐵死亡敏感。結果顯示單獨使用魚藤酮不會誘導PI陽性,而20μM的FAC只導致少量PI陽性細胞(圖5D)。與此相反,當FAC和魚藤酮一起處理P-Hepa時,會導致大量PI 染色,這表明在mtROS產生水平較低的情況下,即使線粒體鐵的少量增加也會引發鐵死亡。
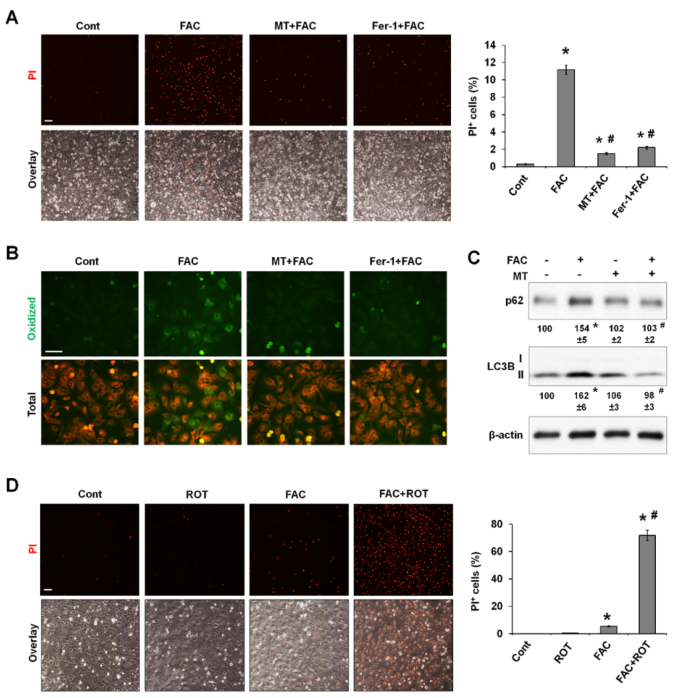
圖5 mtROS的過度生成在鐵誘導的脂質過氧化、自噬功能障礙和鐵死亡中必不可少
4、誘導ATF4保護自噬和細胞存活免受鐵毒性
前面研究結果表明,ATF4可維持自噬和細胞存活,抵御鐵的毒性。為測試ATF4的潛在作用,利用293 T細胞確定沉默ATF4是否會導致自噬和細胞存活失去對鐵毒性的保護。用shATF4質粒構轉染293 T細胞,實現ATF4的敲除(圖6A)。黃色點相對于總數的百分比增加表明自噬通量受損。在對照細胞中,對照條件下黃色點僅占總點數的7%,用FAC處理也沒有引起顯著變化(圖6B)。與此相反,在對照條件下,ATF4基因敲除會降低自噬通量,黃色點的比例為25%,經FAC處理后顯著增加至43%,表明自噬通量進一步降低(圖6B)。同樣,與對照細胞相比,ATF4缺陷細胞會引起LC3B-II的積累,而FAC會增強LC3B-II 的積累(圖6C)。此外,敲除ATF4導致FAC誘導的mtROS生成明顯增加(圖6D)。與對照細胞相比,shATF4細胞對FAC誘導的細胞死亡敏感,而鐵死亡抑制劑Fer-1可減輕這種敏感性(圖6E)。總之,結果表明ATF4在維持自噬功能和防止鐵誘導的mtROS介導的鐵死亡中的作用。

圖6 ATF4缺乏導致鐵依賴性自噬功能障礙、mtROS產生和鐵死亡
5、缺鐵誘導內質網應激和自噬減少
為比較鐵增加和減少的影響,測定缺鐵對不同細胞物種的細胞應激反應和自噬的影響。鐵缺乏是由去鐵胺(DFO)誘導的,并再次測量FTH1的表達,以顯示細胞內的鐵儲存。之前研究表明,AML12和RAW264.7細胞在自噬和細胞存活變化方面對鐵超載不敏感。相反,這兩種細胞系都對缺鐵敏感,表現出ER應激反應,表現為磷酸化ERK、ATF4和CHOP的表達增加以及LC3B的表達增加(圖7A)。這些效應也出現在P-Hepa細胞中,但不出現在HepG2細胞中(圖7B)。氧化應激標志物NRF2的表達在長期缺鐵的情況下有所下降(圖7A)。在AML12細胞中使用自噬抑制劑Chlq,繼續評估缺鐵如何調控自噬通量。DFO再次誘導LC3B-II表達的顯著增加,但與單獨使用Chlq相比,DFO/Chlq聯合處理并沒有進一步增加LC3B-II(圖7C),這表明缺鐵時自噬通量減少。此外,DFO處理的AML12細胞中裂解的Caspase-3(CC3)表達增加,表明長期缺鐵導致細胞凋亡(圖7D)。
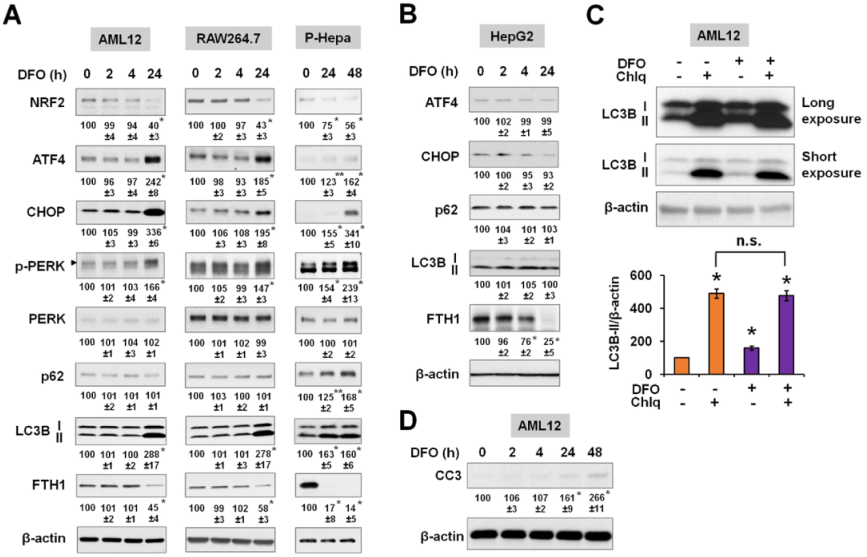
圖7鐵缺乏可誘導內質網應激反應并降低自噬通量
通過使用腸上皮特異性鐵輸出蛋白(Fpn)基因敲除小鼠(FpnΔIEC)確定體內缺鐵對肝臟ER應激反應和自噬的影響。與之前的報道一致,FpnΔIEC小鼠在Fpn基因敲除6周后出現生長遲緩和明顯貧血(腳趾呈灰色)(圖8A)表現為FTH1的表達明顯降低(圖8B)。與對照組Fpnf/f小鼠相比,FpnΔIEC小鼠p-ERK和CHOP的表達明顯升高(圖8B),表明ER壓力增加。缺鐵肝臟中CC3的表達也增加(圖8B)。同樣,熒光染色顯示FpnΔIEC小鼠肝臟中Grp78和CC3的表達增加(圖8C)。與DFO誘導的培養細胞急性缺鐵不同,長期缺鐵的FpnΔIEC肝臟中p62和LC3B蛋白表達量顯著下降(圖8B)。文獻表明ER應激可誘導轉錄重編程,導致許多基因的轉錄抑制。事實上,FpnΔIEC肝臟中的LC3和p62轉錄本明顯減少(圖8D),這也是LC3B和p62蛋白減少的原因。此外,Chlq使對照組肝臟中的 LC3B-II 表達量明顯增加了65%,但在FpnΔIEC肝臟中卻沒有引起任何明顯變化(圖8E)。同樣,Chlq可使對照組肝臟中p62的表達增加90%,但在FpnΔIEC肝臟中僅增加43%。LC3B-II和p62在Chlq作用下的積累減少表明缺鐵肝臟的自噬通量下降。
缺鐵誘導的ER應激可通過TEM分析再現,在FpnΔIEC肝細胞中可看到輕微擴張的ER(圖8F)。在FpnΔIEC 肝細胞中看到糖原(紅色箭頭)和脂滴(藍色箭頭)堆積增加(圖8F)。與之前的報告一致,缺鐵的FpnΔIEC肝細胞中線粒體明顯增大(圖8G),引人注目的是,與野生型肝細胞相比,FpnΔIEC 肝細胞中溶酶體的數量明顯減少(圖8H),這至少是自噬通量減少的部分原因。
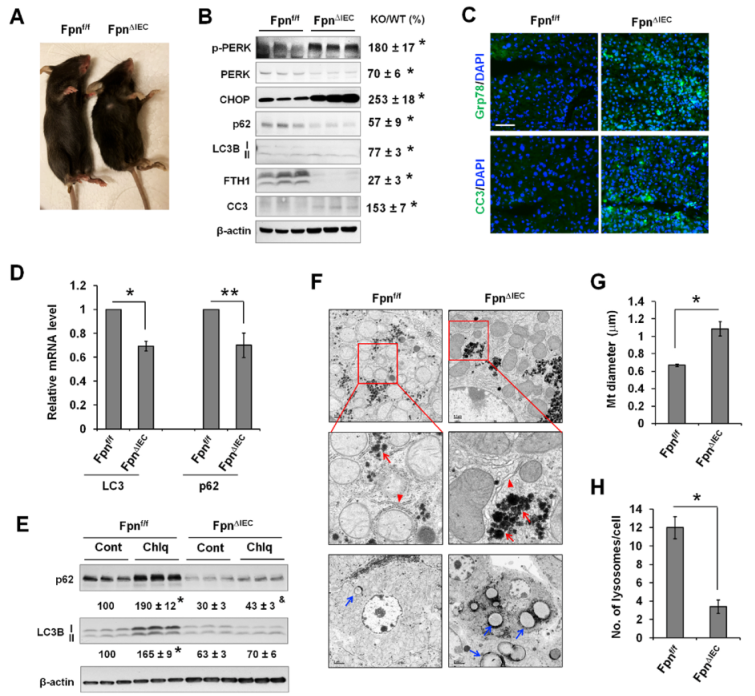
圖8鐵缺乏誘導內質網應激,并通過減少體內肝臟溶酶體生物發生而降低自噬
6、鐵缺乏引起的自噬通量降低依賴于ER應激的誘導
作者試圖確定與缺鐵相關的ER平衡紊亂是如何導致自噬通量下降的。LAMP1是一種糖基化蛋白質,是溶酶體生物生成和自噬不可或缺的組成部分。結果顯示ER應激的DFO和曲卡霉素,兩種通過抑制蛋白N-糖基化的自噬誘導劑,都降低LAMP1的成熟(圖9A)。4-PBA能幫助蛋白質折疊并抑制ER應激,它能抑制DFO誘導的Grp78表達并改善LAMP1的成熟(圖9B)。重要的是,4-PBA可減輕DFO誘導的LC3B-II的積累。
作者推測溶酶體蛋白的成熟缺陷會損害溶酶體的生物生成。對溶酶體蛋白酶 Cathepsin B進行染色[50],以檢測溶酶體數量的潛在變化。LysoTracker沒有被使用,因為它既能標記溶酶體,也能標記晚期核內體。DFO處理導致cathepsin B 陽性點的數量明顯減少,而與4-PBA聯合處理可部分緩解這一現象(圖9C)。通過使用自噬通量報告基因GFP-LC3-RFP,進一步發現,DFO誘導的黃色點狀突起的百分比顯著增加,而與4-PBA共同處理后,黃色點狀突起的百分比顯著減少(圖9D),這表明蛋白質成熟的恢復和ER應激的抑制改善了自噬通量。值得注意的是,與未處理的對照組相比,4-PBA 本身也會導致黃色點的百分比輕微但顯著地增加。總之,結果表明,缺鐵供應擾亂了ER平衡和蛋白質成熟,至少是溶酶體生物生成受損和自噬減少的部分原因。
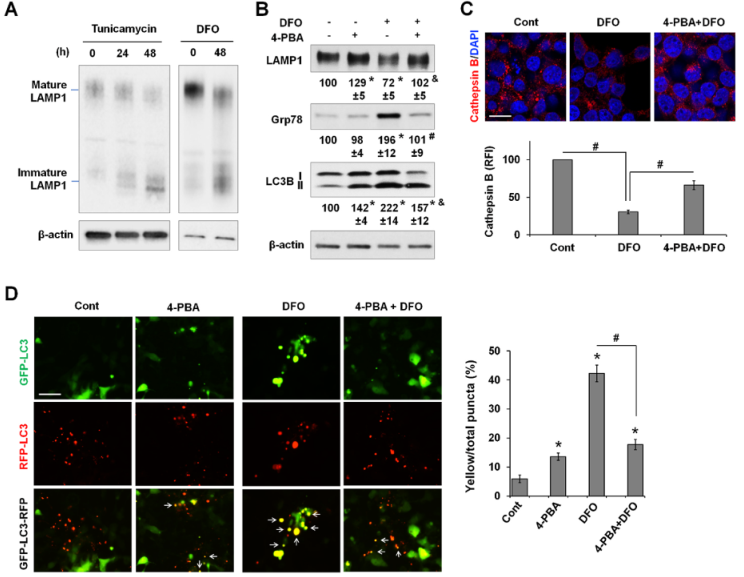
圖9缺鐵導致的自噬減少與內質網應激有關
總之,本研究表明,一方面鐵過量會誘導產生mtROS,從而引起脂質過氧化反應依賴性鐵死亡和自噬通量抑制。自噬的減少進一步加強了鐵過量誘導的鐵死亡。ATF4的上調以及使用mtROS清除劑MT或過氧化脂質清除劑Fer-1可阻礙脂質過氧化和自噬通量的受損,從而阻止鐵過量誘導的鐵死亡。另一方面,缺鐵會損害蛋白質折疊和成熟,導致ER應激和溶酶體生物生成減少,共同導致自噬通量下降。4-PBA可改善蛋白質的成熟并減輕ER壓力,從而挽救溶酶體的生物生成和自噬通量。推測ER應激和自噬通量受損都有助于誘導細胞凋亡。
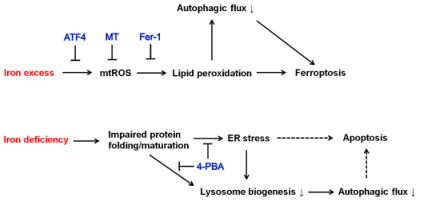
圖10 鐵過載和鐵缺乏對細胞壓力響應,自噬,細胞存活影響的示意圖
實驗方法
qRT-PCR,WB,PI染色檢測細胞死亡,共聚焦免疫熒光,ROS,Fe2+,線粒體膜電位,脂質過氧化實驗,透射電子顯微鏡,自噬通量
參考文獻
Wang Y, Wang M, Liu Y, Tao H, Banerjee S, Srinivasan S, Nemeth E, Czaja MJ, He P. Integrated regulation of stress responses, autophagy and survival by altered intracellular iron stores. Redox Biol. 2022 Jul 14;55:102407. doi: 10.1016/j.redox.2022.102407.