






METTL16促進糖酵解代謝重編程和結直腸癌進展
糖酵解是癌癥的關鍵標志,維持惡性腫瘤的發生和發展。m6A修飾在糖酵解中的作用在很大程度上是未知的。本研究探討了m6A甲基轉移酶METTL16在糖酵解代謝中的生物學功能,揭示了結直腸癌(CRC)進展的新機制。研究發現,SOGA1是METTL16的直接下游靶點,參與METTL16介導的糖酵解和CRC進展。METTL16通過結合IGF2BP1顯著增強SOGA1的表達和mRNA的穩定性。隨后,SOGA1促進AMPK復合體泛素化,抑制其表達和磷酸化,從而上調PDK4。此外,YY1可以通過直接結合METTL16的啟動子,通過轉錄抑制其在CRC細胞中的表達。臨床資料顯示,METTL16表達與SOGA1、PDK4呈正相關,與結直腸癌患者預后不良有關。我們的研究結果表明METTL16/SOGA1/PDK4軸可能是CRC的有希望的治療靶點。本文于2023年6月發表于“Journal of Experimental & Clinical Cancer Research”(IF=11.3)上。
技術路線

結果
1)METTL16過表達與結直腸癌患者預后不良相關
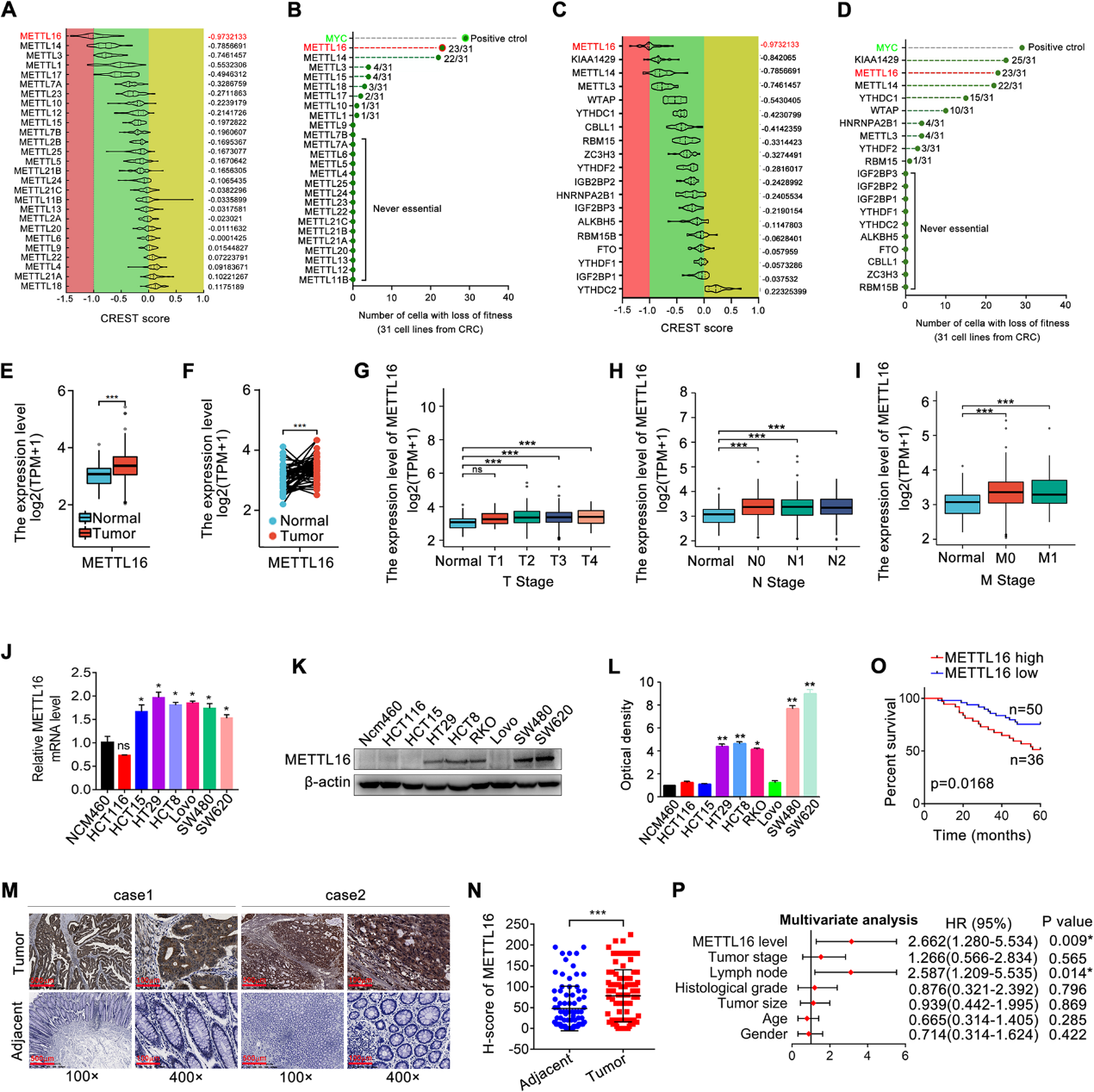
通過對數據集的分析,我們發現在METTL家族成員中,METTL16是CRC細胞存活最重要的基因(圖1A, B)。重要的是,在m6A的主要調控因子中,METTL16也在CRC的存活中顯示出重要作用(圖1C, D),說明其在CRC中的功能意義。TCGA和GEO數據庫顯示,與正常組織相比,METTL16在結直腸癌組織中顯著上調(圖1E、F)。此外,TGGA數據顯示與METTL16表達和CRC的臨床病理變量相關。如圖1G-I所示,METTL16表達升高與腫瘤大小、淋巴結轉移、遠處轉移及臨床分期分級顯著相關。METTL16 mRNA在結直腸癌細胞系中的表達也高于正常結腸上皮細胞(圖1J)。同樣,METTL16蛋白在結直腸癌細胞系中的表達也普遍高于正常結腸上皮細胞(圖1K、L)。免疫組化(IHC)對結直腸癌樣本和癌旁組織的染色結果進一步證實了METTL16在結直腸癌組織中的表達增強(圖1M和N)。METTL16蛋白表達的增加與結直腸癌患者的低生存率相關(圖1O)。此外,多變量Cox回歸分析顯示,METTL16蛋白表達可能是結直腸癌患者生存的獨立預測因子(圖1P)。總的來說,METTL16在CRC中上調,可能在CRC進展中發揮重要作用。
2)METTL16促進CRC進展
為了研究METTL16在結直腸癌進展中的作用,我們分別使用兩種shRNA (shM161、shM16-2)和pHBLV-METTL16載體(OEM16)在結直腸癌細胞中敲低和過表達METTL16的表達。敲低METTL16可降低CRC細胞的增殖和集落形成,而過表達METTL16則具有相反的作用(圖2A-D)。同樣,METTL16敲低抑制了CRC細胞的遷移和侵襲能力,而METTL16過表達促進了CRC細胞的轉移(圖2E、F)。在METTL16敲低和過表達的CRC異種移植物中進一步評估了METTL116的功能,結果表明METTL116促進腫瘤生長,而抑制METTL16則抑制腫瘤生長,這可以通過腫瘤的大小、體積和重量反映出來(圖2G-L)。此外,METTL16敲低降低了Ki67的表達,而METTL16過表達則促進了Ki67在體內的表達(圖2M, N)。綜上所述,這些結果表明METTL16在促進CRC進展中發揮了重要作用。

3)SOGA1是METTL16的直接靶點
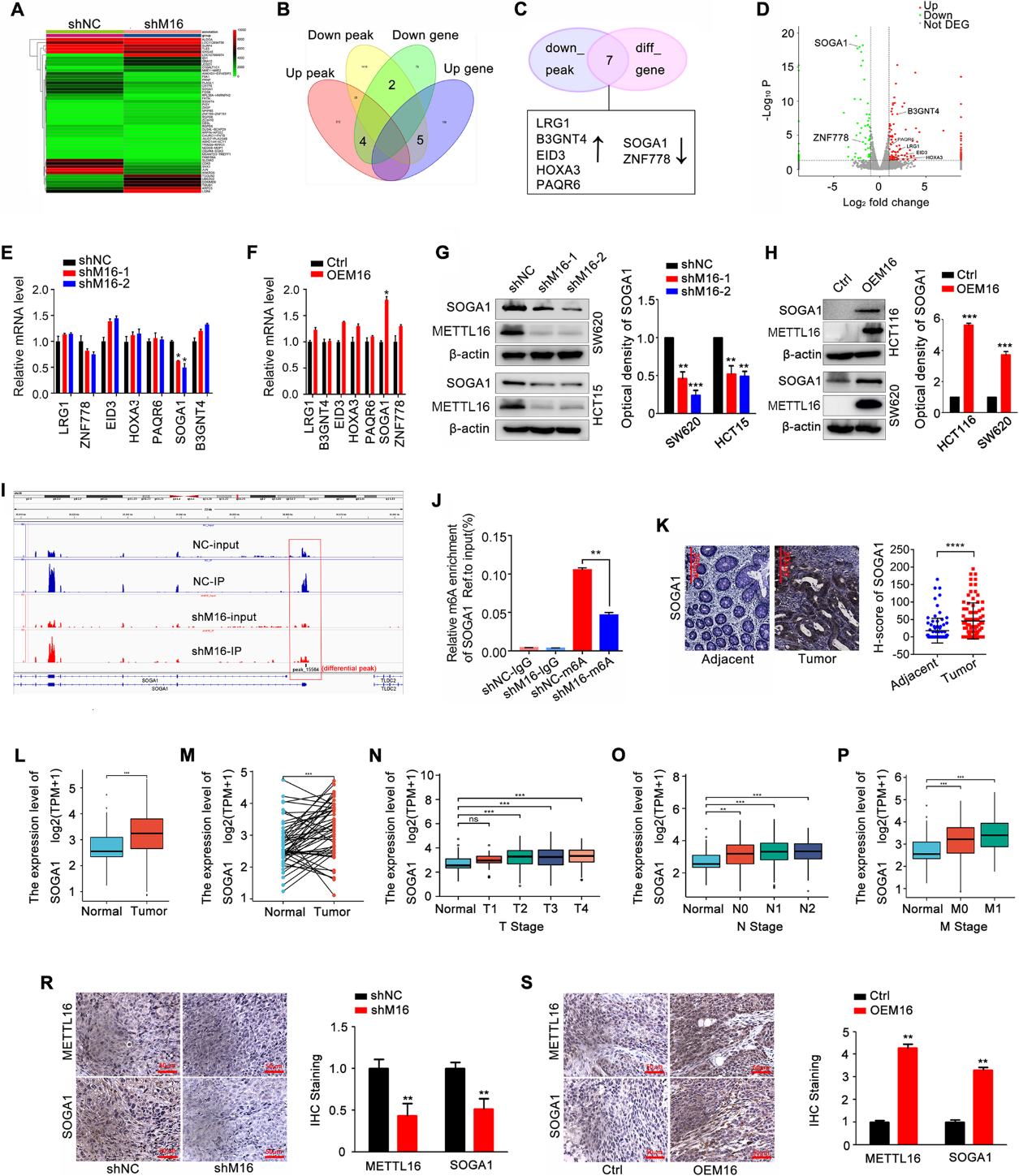
為了探究METTL16誘導CRC增殖的分子機制,我們對METTL16穩定敲除的CRC細胞和對照細胞進行了MeRIP測序和RNA測序。圖3A顯示了前50個差異基因,9個變化基因和峰重疊(圖3B)。在低峰中,有7個基因表達改變,包括LRG1, B3GNT4, EID3, HOXA3, PAQR6, SOGA1和ZNF778(圖3C, D)。通過驗證,在結直腸癌細胞中,METTL16的敲低降低了SOGA1 mRNA的表達,而METTL16的過表達上調了SOGA1 mRNA的表達(圖3E, F)。同樣,METTL16正調控SOGA1蛋白的表達(圖3G, H)。重要的是,在MeRIP-seq數據中,我們檢測到SOGA1 mRNA的一個m6A峰,該峰在METTL16敲除后減弱(圖3I)。MeRIP-qPCR結果顯示,當METTL16被敲除時,m6A修飾的SOGA1 mRNA顯著減少(圖3J)。作為致癌基因,SOGA1蛋白和mRNA在結直腸癌組織中的表達與正常組織相比顯著升高(圖3K-M)。SOGA1表達升高與腫瘤大小、淋巴結轉移、遠處轉移及臨床分期分級顯著相關(圖3N-P)。此外,METTL16在體內正調控SOGA1蛋白表達(圖3R, S)。綜上所述,這些結果表明SOGA1是METTL16的直接靶點。
4)IGF2BP1是SOGA1的m6A閱讀器
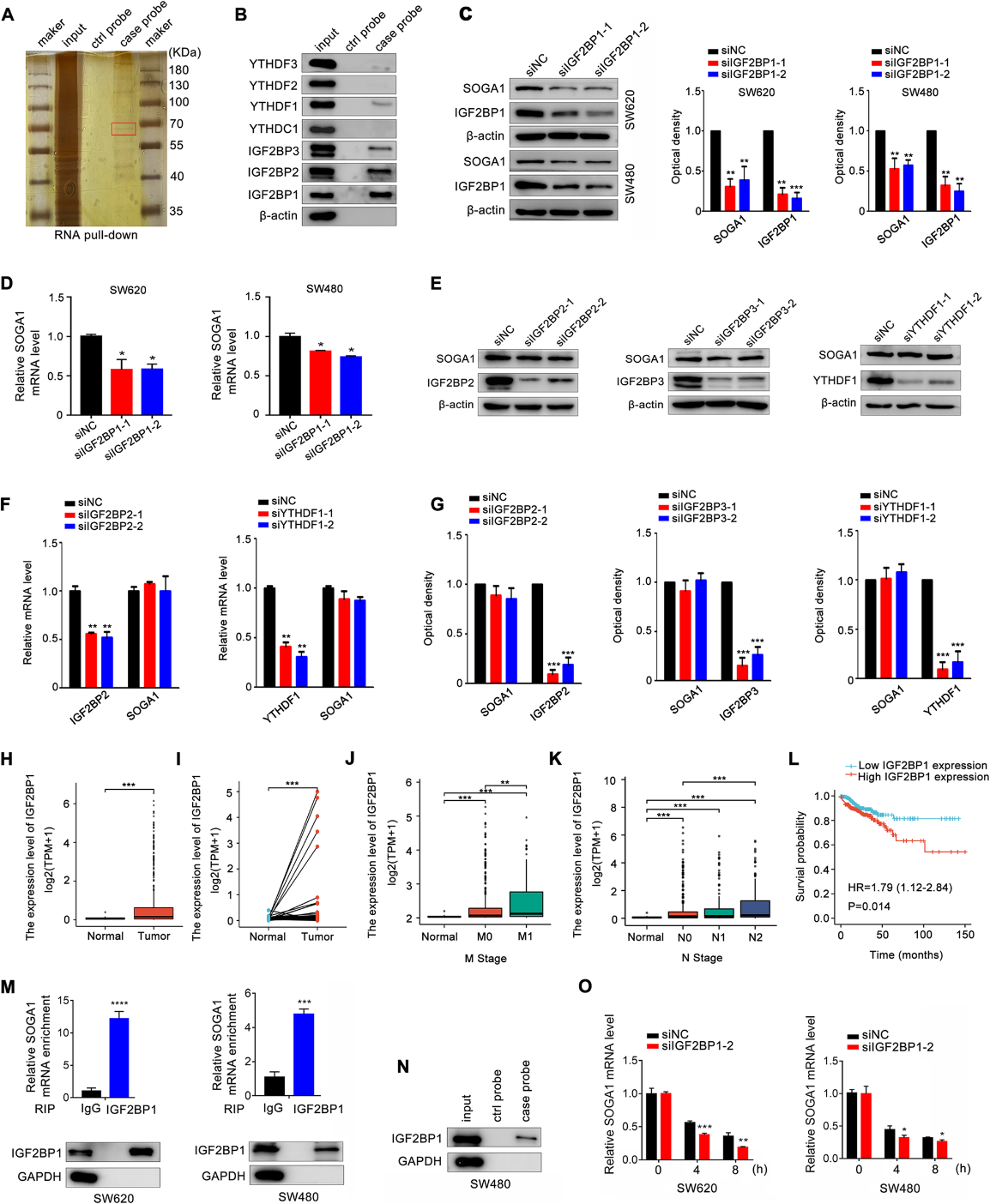
我們進一步研究了m6A修飾SOGA1 mRNA的機制。為了鑒定識別和結合SOGA1甲基化的m6A閱讀器,我們進行了RNA下拉實驗,從SW620細胞中捕獲SOGA1相互作用的閱讀器。m6A閱讀器YTHDF1和IGF2BP1/2/3與SOGA1 mRNA結合(圖4A, B)。有趣的是,敲低IGF2BP1可顯著下調結直腸癌細胞中SOGA1 mRNA和蛋白水平(圖4C, D)。然而,敲低YTHDF1和IGF2BP2/3對SOGA1蛋白表達無明顯影響(圖4E-G)。TCGA數據分析顯示,與正常組織相比,IGF2BP1在結直腸癌組織中表達上調(圖4H、I), IGF2BP1表達升高與腫瘤大小、淋巴結轉移、遠處轉移和臨床分期分級顯著相關(圖4J、K)。IGF2BP1表達升高與CRC患者生存不良相關(圖4L)。RNA下拉實驗揭示了IGF2BP1蛋白與SOGA1 mRNA之間的密切相互作用(圖4M)。同樣,RIP實驗的結果進一步證實了IGF2BP1直接與SOGA1 mRNA結合(圖4N)。IGF2BP1敲低降低了SOGA1 mRNA的穩定性,提高了其在CRC細胞中的降解率(圖4O)。綜上所述,這些結果表明甲基化的SOGA1 mRNA被IGF2BP1直接識別,從而抑制轉錄物降解并以依賴m6A的方式促進SOGA1的表達。
5)METTL16/SOGA1通過調節PDK4的表達促進糖酵解
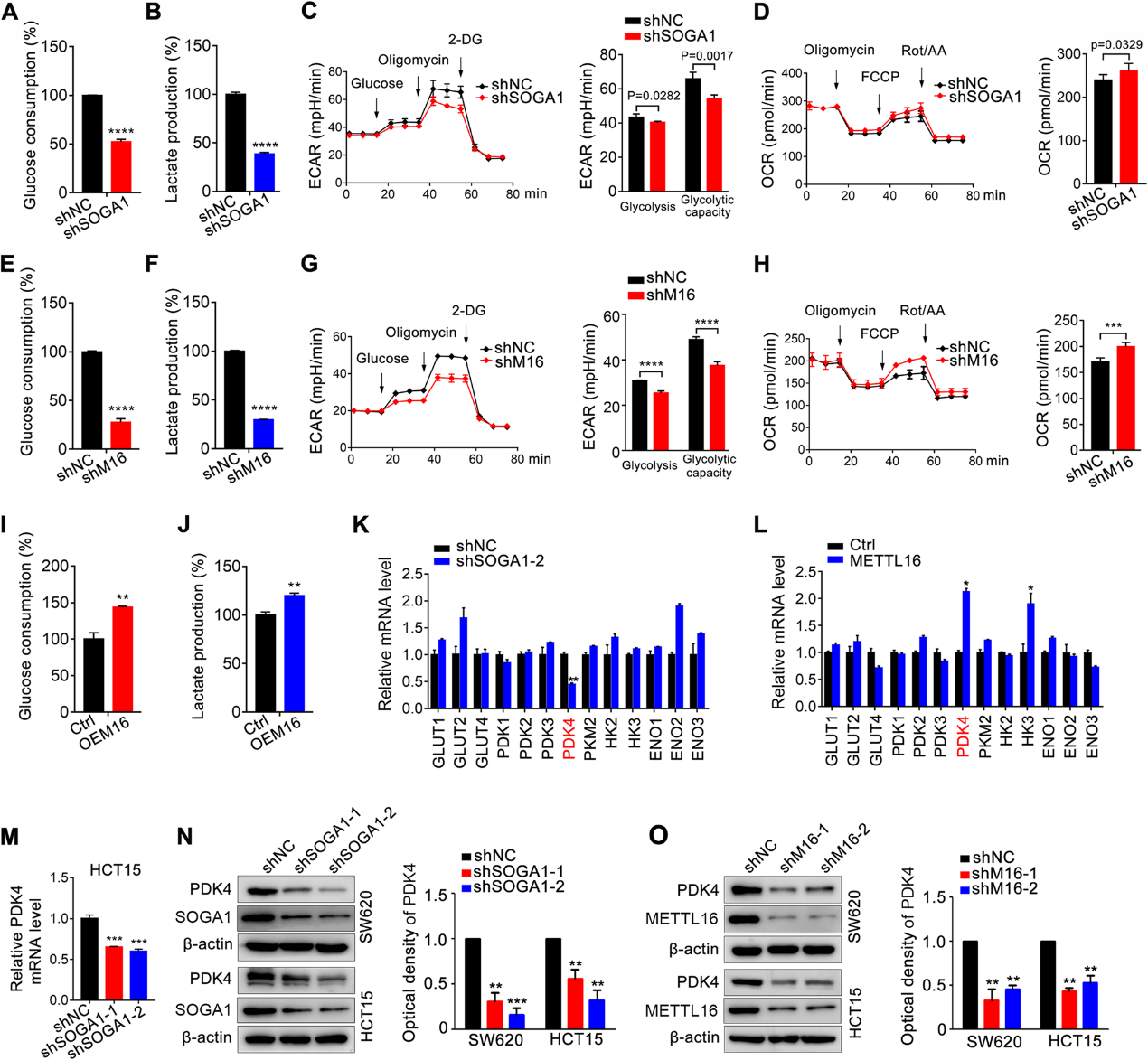
我們研究了METTL16/ SOGA1軸在糖酵解中促進CRC進展的作用。SW620細胞中SOGA1的下調顯著降低了葡萄糖攝取和乳酸生成(圖5A, B)。此外,SOGA1缺失的CRC細胞顯示ECAR降低,OCR增加,反映了線粒體氧化呼吸(圖5C, D)。與此一致,METTL16缺失的CRC細胞顯示葡萄糖攝取(圖5E)、乳酸生成(圖5F)、ECAR(圖5G)降低,OCR增加(圖5H)。與此相反,SW620細胞中METTL16的過表達促進了葡萄糖攝取和乳酸生成(圖51,J)。為了進一步確定METTL16/SOGA1介導糖酵解的機制,我們測量了SOGA1敲低CRC細胞中一系列葡萄糖代謝相關基因的mRNA表達(圖5K)。我們還研究了這些分子是否受METTL16的控制。因此,我們檢測了這些基因在過表達的METTL16 crc細胞中的mRNA表達(圖5L)。有趣的是,只有PDK4的表達水平因SOGA1的下調而顯著降低,而因METTL16的過表達則持續升高。通過進一步驗證,我們發現SOGA1和METTL16的敲低降低了CRC細胞中PDK4 mRNA和蛋白的表達(圖5M-O)。總的來說,我們的數據顯示METTL16/SOGA1軸通過調節CRC細胞中PDK4的表達來促進糖酵解。
6)SOGA1通過抑制AMPK信號傳導促進PDK4的表達
我們進一步探討了SOGA1調控PDK4表達的潛在機制。有報道稱AMPK信號是PDK4上游的關鍵信號。為了驗證AMPK在PDK4表達中的作用,我們使用AMPK激活劑A769662刺激CRC細胞。結果表明,A769662誘導AMPK (Thr172)磷酸化,激活AMPK信號,顯著降低PDK4蛋白和mRNA的表達(圖6A、B)。我們發現,AMPKα1、β1和γ1亞基的敲低增加了PDK4 mRNA和蛋白的表達(圖6C、D),表明AMPK位于PDK4的上游,負向調節CRC細胞中PDK4的表達。接下來,我們發現敲低SOGA1顯著促進AMPK磷酸化和AMPKα1、β1和γ1蛋白表達(圖6F),但對AMPKα1、β1和γ1 mRNA表達無明顯影響(圖6E)。蛋白穩定性分析顯示,SOGA1敲低可降低pAMPK、AMPKα1、β1和γ1的衰變,增強其蛋白穩定性(圖6G)。Co-IP實驗結果顯示,SOGA1可以結合pAMPK、AMPKα1、β1、γ1(圖6H),促進AMPKα1、β1、γ1和pAMPK泛素化(圖6I)。此外,抑制AMPKα1、β1和γ1部分逆轉SOGA1或METTL16的缺失下調PDK4的表達(圖6J)。這些結果表明,SOGA1結合AMPKα1、β1和γ1,誘導其泛素化,抑制其表達和磷酸化,從而促進PDK4的表達(圖6K)。
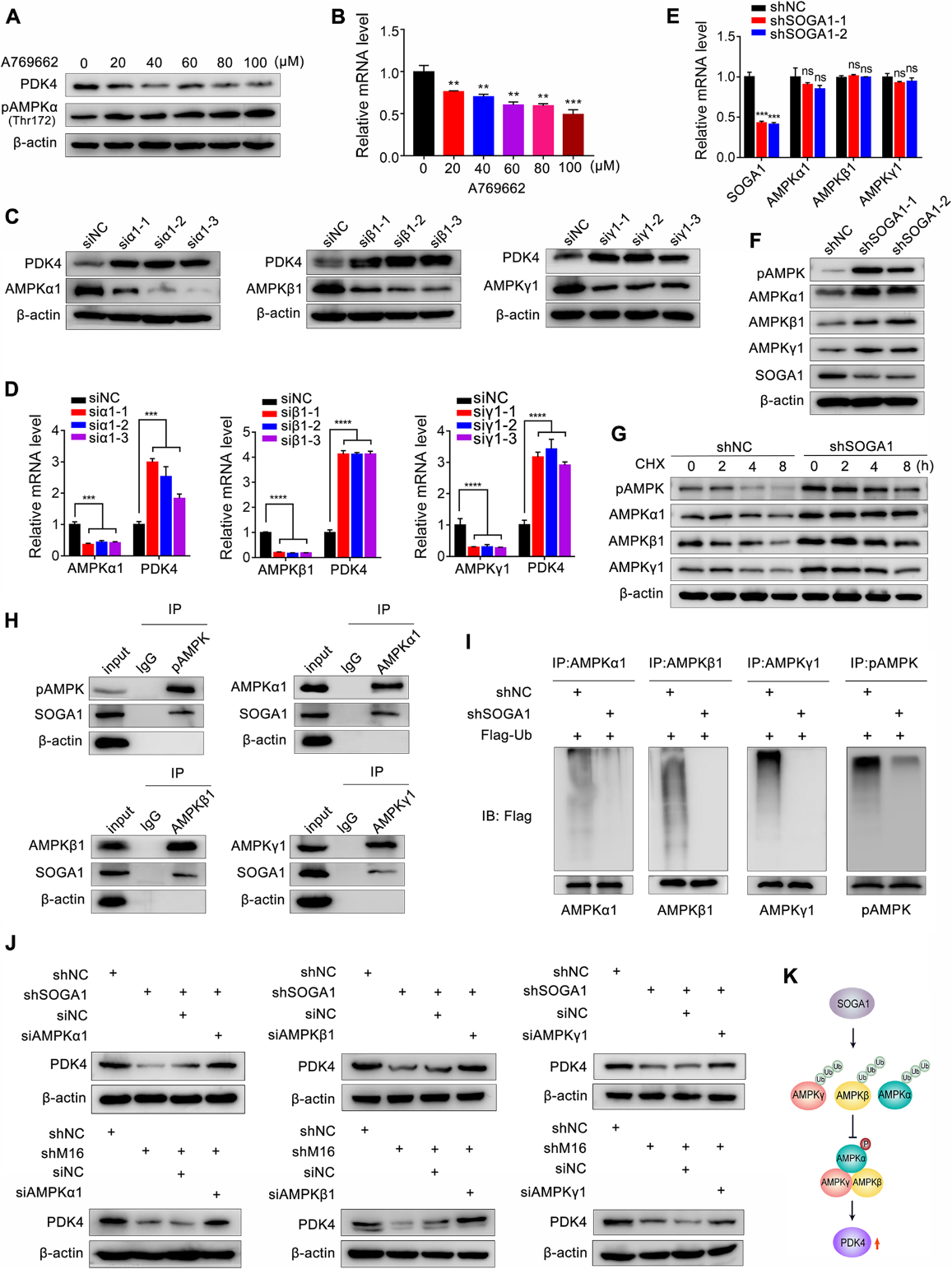
7)YY1通過直接結合METTL16啟動子下調其表達
為了探索METTL16在CRC中高表達的潛在機制,我們通過分析ChIPBase和PROMO中的ENCODE染色質免疫沉淀測序(ChIP-seq)數據,評估了負責調節METTL16的潛在轉錄因子(TFs)。如圖7A所示,CEBPB、YY1、VDR、ETS1、TCF4、XBP1 6個TF重疊在ChIPBase預測的26個TF和PROMO預測的46個TF中。接下來,分別下調這6個TF,發現CEBPB和YY1的敲低增加了METTL16 mRNA的表達(圖7B)。然而,CEBPB的缺失對METTL16蛋白的表達沒有明顯影響(圖7C)。YY1的敲低明顯上調了CRC細胞中METTL16蛋白和mRNA的表達(圖7D, E)。在對METTL16基因啟動子的分析中,我們找到了YY1在其上的結合位點,并設計了ChIP引物(圖7F)。ChIP實驗表明,YY1可以直接結合CRC細胞中的METTL16啟動子(圖7G),表明YY1是METTL16的上游TF。接下來,我們對METTL16啟動子報告基因的兩個YY1潛在結合位點進行突變,生成pGL-M16-Mut1或pGL-M16-Mut2(圖7H)。結果表明,si-YY1可以顯著降低pGLM16-WT和pGL-M16-Mut1的熒光素酶水平,而si-YY1對pGL-M16-Mut2的抑制作用減弱(圖7I)。此外,YY1敲低增加了CRC細胞中SOGA1 mRNA的穩定性(圖7J)。綜上所述,這些數據表明YY1是METTL16的上游轉錄因子,并通過直接結合METTL16的啟動子來調節METTL16的表達。
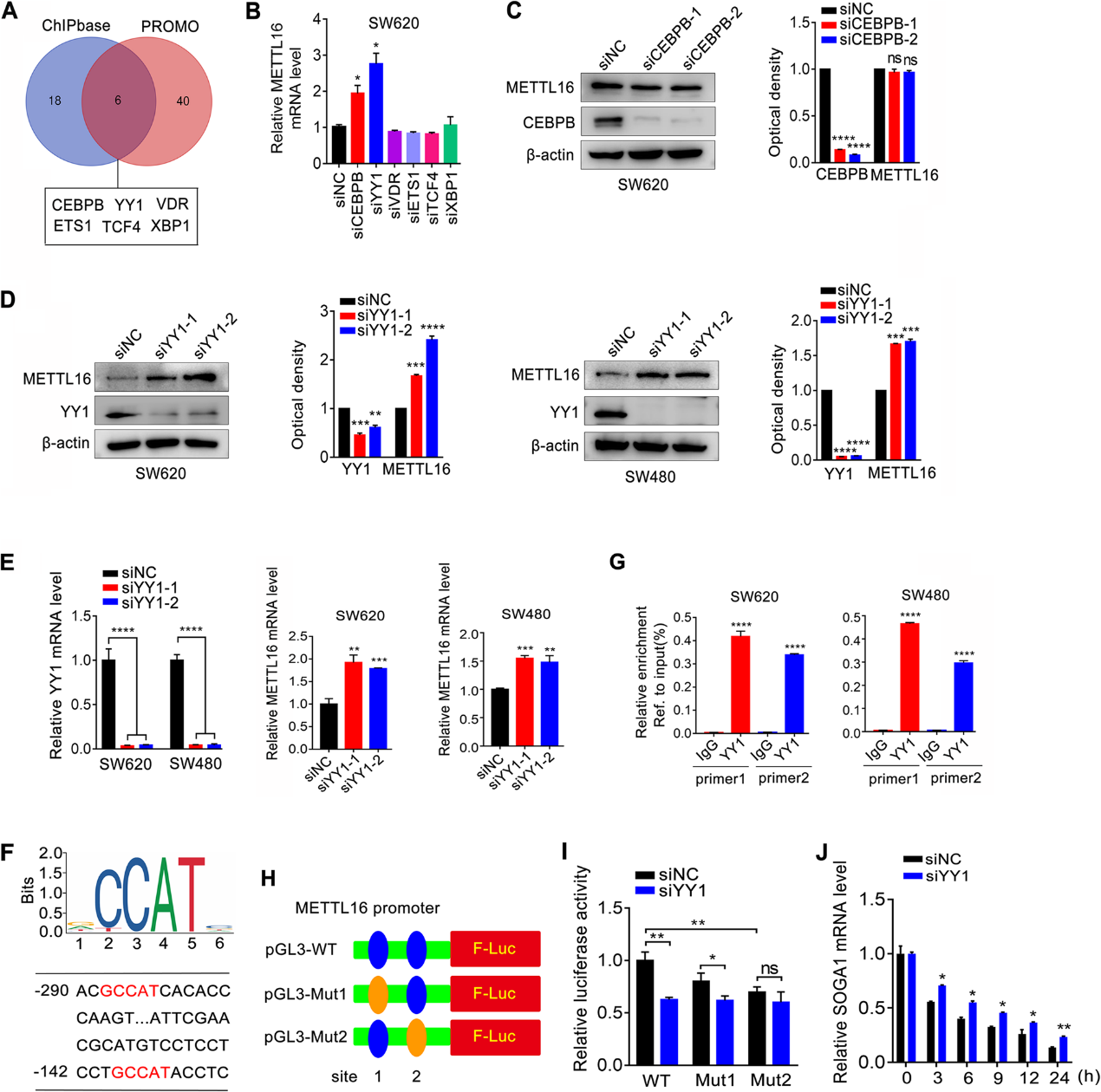
8)METTL16/SOGA1軸與結直腸癌患者預后不良有臨床相關性
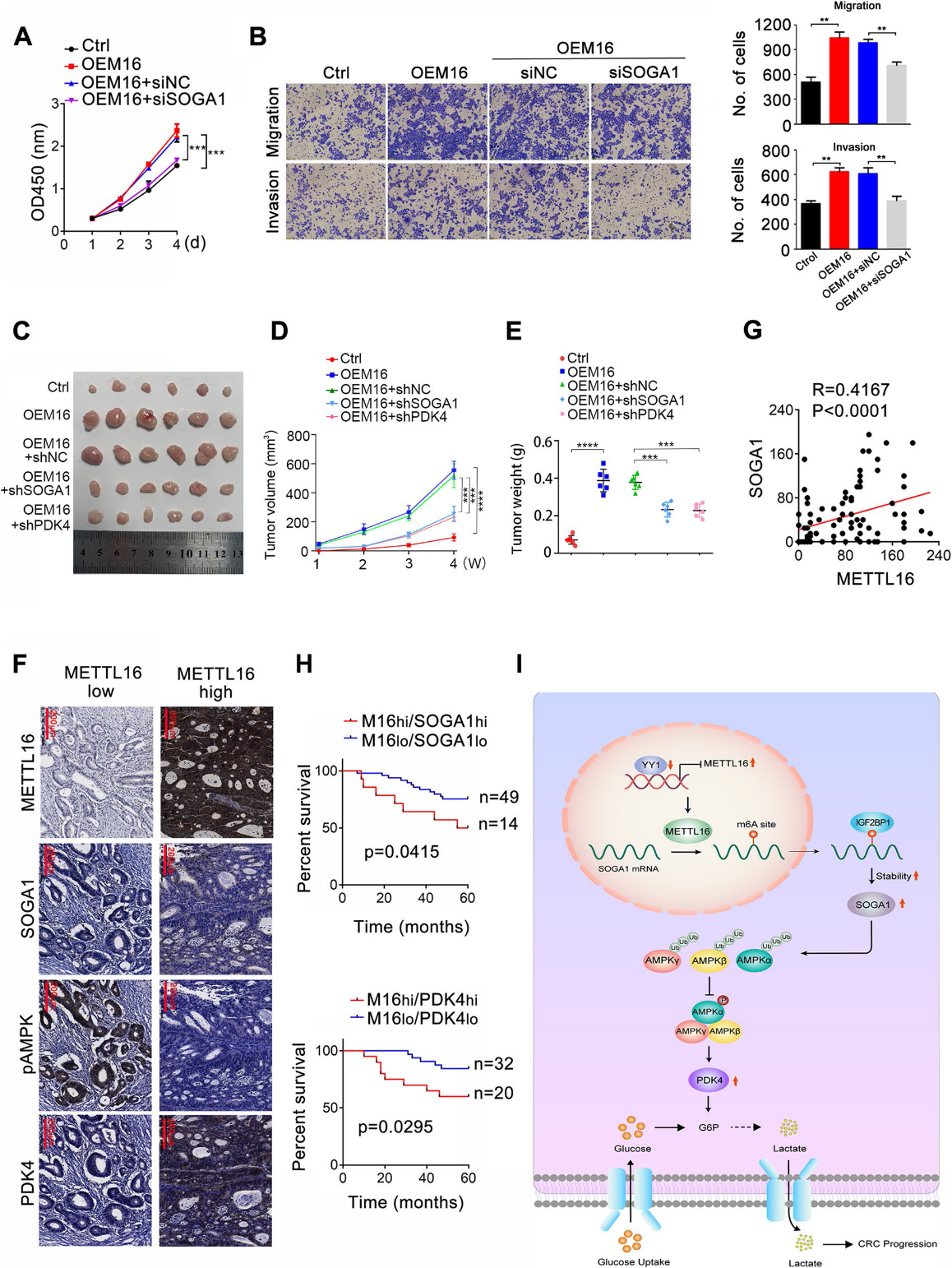
為了研究SOGA1在METTL16介導的CRC增殖中的作用,我們在METTL16過表達的CRC細胞中使用siRNA下調SOGA1的表達并檢測細胞增殖。結果表明,SOGA1的敲低削弱了METTL16促進的CRC細胞的增殖(圖8A)。SOGA1敲低部分抑制了METTL16促進的CRC細胞轉移(圖8B)。在體內,SOGA1和PDK4敲低都減弱了METTL116的過表達提高的腫瘤生長,這反映在腫瘤大小(圖8C)、體積(圖8D)和重量(圖8E)上。此外,METTL16和SOGA1在結直腸癌組織中的表達呈正相關(圖8F、G)。此外,在結直腸癌組織中,METTL16的表達與PDK4呈正相關(圖8F)。重要的是,Kaplan-Meier分析顯示,METTL16與SOGA1共表達或METTL16與PDK4高表達與CRC患者預后不良呈正相關(圖8H)。上述結果表明,SOGA1在METTL16介導的CRC細胞增殖中起重要作用,METTL16/SOGA1軸與CRC患者預后不良具有臨床相關性。
結論
我們的研究結果表明METTL16在結直腸癌進展中具有腫瘤促進作用。METTL16在結直腸癌組織中表達上調,與結直腸癌患者預后不良相關。機制上,METTL16/SOGA1/ PDK4信號軸通過誘導糖酵解促進結直腸癌的進展。這一發現為探索結直腸癌新的診斷生物標志物和治療靶點提供了新的見解。
實驗方法
Western blotting,qRT?PCR,代謝試驗,ECAR和OCR,RNA穩定性和蛋白質穩定性測定,Co?IP,IHC,transwell,MeRIP測序,RIP,RNA pull?down,ChIP,動物實驗。
參考文獻
Wei W, Zhang ZY, Shi B, Cai Y, Zhang HS, Sun CL, Fei YF, Zhong W, Zhang S, Wang C, He B, Jiang GM, Wang H. METTL16 promotes glycolytic metabolism reprogramming and colorectal cancer progression. J Exp Clin Cancer Res. 2023 Jun 20;42(1):151. doi: 10.1186/s13046-023-02732-y.