






ZBP1可預防mtDNA誘導的衰竭心臟的心肌炎癥
欄目:最新研究動態
發布時間:2023-08-23
ZBP1(Z-DNA 結合蛋白1)是一種模式識別受體,可響應炎癥細胞、成纖維細胞和內皮細胞中的mtDNA 而積極調節炎癥......

心力衰竭(HF)的患病率和發病率正在上升,并被定義為一種全球大流行,影響全球約26萬人。盡管治療取得了重大進展,但HF的發病率和死亡率仍然很高。因此,需要對HF的病理生理學和分子機制有新的見解來開發新的治療方法。線粒體DNA(mtDNA)誘導的心肌炎癥與心臟重塑密切相關。ZBP1(Z-DNA 結合蛋白1)是一種模式識別受體,可響應炎癥細胞、成纖維細胞和內皮細胞中的mtDNA 而積極調節炎癥。然而,ZBP1在心肌炎癥和心臟重塑中的作用尚不清楚。該研究發表于《Circulation Research》,IF:23.213。
技術路線:

主要研究結果:
1. mtDNA增加ZBP1并激活心肌細胞中的炎癥信號傳導
為了評估心肌細胞中ZBP1對mtDNA的表達,作者將1000ng / mL從大鼠肝臟中提取的mtDNA施用到心肌細胞中,通過檢測mtDNA的一部分Cox1而不是核DNA的一部分At3來驗證其純度(圖1A)。從mtDNA給藥24小時后,作者發現ZBP1 mRNA和蛋白質水平增加(圖1B和1C)隨著IL-1β和IL-6 mRNA水平的增加(圖1D)。
此外,作者研究了ZBP1和其他與炎癥相關的蛋白質的mtDNA劑量依賴性反應,包括微管相關蛋白1A / 1B- LC3(輕鏈3),RIPK3,磷酸化的NF-κB p65亞基(Ser536)和NLRP3。雖然低劑量的mtDNA(100 ng/mL)增加了LC3-II,但沒有增加ZBP1,但高劑量的mtDNA(1000 ng/mL)增加了LC3-II和ZBP1(圖1E和1F)。重要的是,只有高劑量的mtDNA才能增加RIPK3,磷酸化的NF-κB,NLRP3和磷酸化TBK1的蛋白質水平,它們是炎癥的主要介質(圖1F)。此外,高劑量的mtDNA增加了IL-1β和IL-6的mRNA水平,而低劑量則沒有(圖1G)。這些數據表明,雖然低劑量的mtDNA通過自噬激活和處理,如LC3-II的增加所證明的那樣,高劑量的mtDNA不僅誘導自噬,而且還增加ZBP1和其他炎癥相關蛋白,導致心肌炎癥。
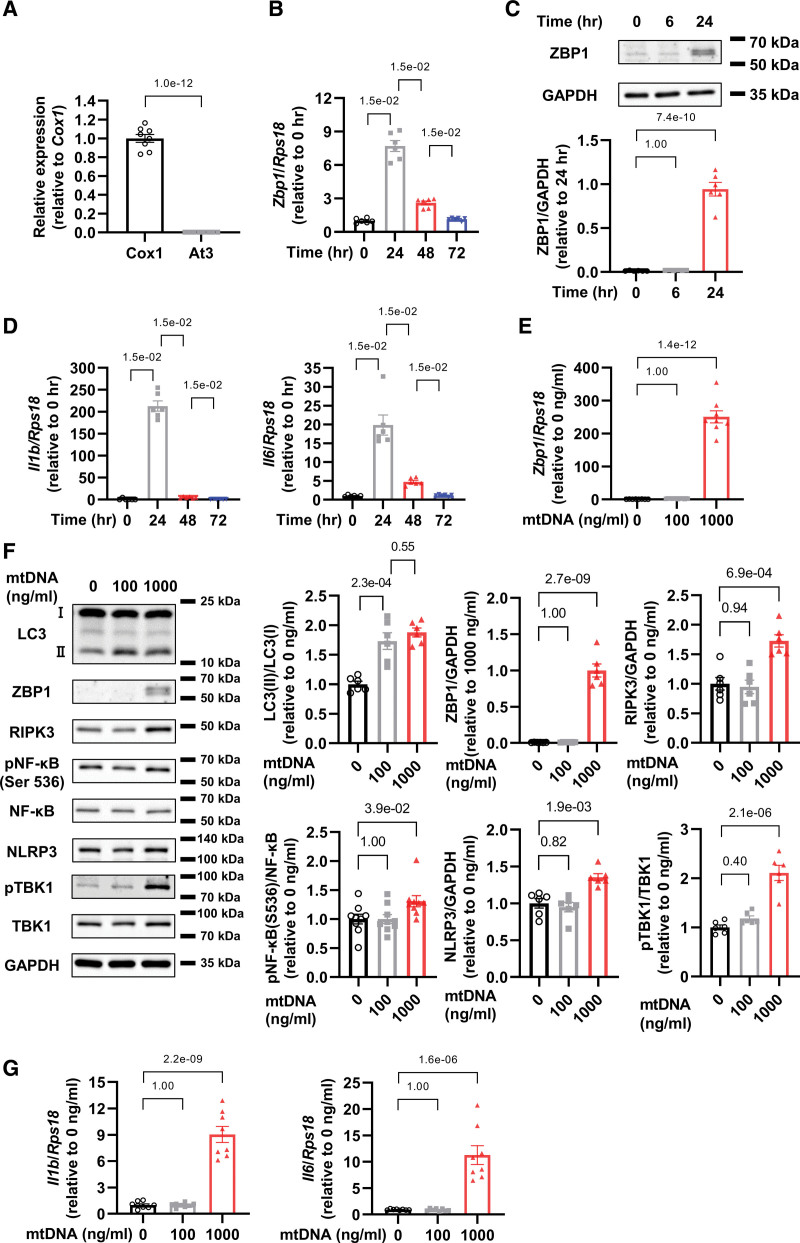
圖1 mtDNA增加ZBP1并激活心肌細胞中的炎癥信號傳導
2. ZBP1敲低加劇了用mtDNA處理的心肌細胞中炎癥細胞因子的增加
接下來,作者研究了ZBP1在心肌細胞中響應1000 ng / mL mtDNA的功能作用。小干擾RNA敲低有效抑制了ZBP1的mRNA和蛋白質水平(圖2A和2B)。出乎意料的是,盡管ZBP-1已被報道介導炎癥,但ZBP1敲低加劇了mtDNA誘導的RIPK3,磷酸化NF-κB和NLRP3的增加(圖2B)。與這些結果一致,ZBP1敲低進一步增加了用mtDNA處理的心肌細胞中IL-1β和IL-6的mRNA水平(圖2C)。TBK1是ZBP1的另一個下游靶標。盡管高劑量的mtDNA增加了TBK1的磷酸化(圖1F),ZBP1敲低沒有影響它(圖2D)。這些發現表明,ZBP1負調節心肌細胞中的RIPK3,NF-κB和炎性細胞因子,但不是TBK1。

圖2 ZBP1敲低會加劇用mtDNA處理的心肌細胞中炎性細胞因子的增加
3. 在mtDNA處理的心肌細胞中,敲低RIPK3可通過敲低ZBP1抵消NF-κB-NLRP3軸的增加
為了研究ZBP1和RIPK3之間的關系,并闡明RIPK3是否可以介導炎癥作為ZBP1的下游靶標,作者進行了ZBP1和RIPK3的雙重敲低實驗。雙敲低有效降低了這些蛋白質(圖3A)。重要的是,RIPK3敲低減弱了mtDNA處理的心肌細胞中ZBP3敲低磷酸化NF-κB和NLRP1增加的惡化(圖3A)。RIPK3敲低也降低了IL-1β和IL-6的mRNA水平(圖3B)。這些發現表明RIPK3正調控NF-κB和NLRP3,ZBP1通過抑制RIPK3發揮抗炎作用。相反,mtDNA和ZBP1敲低都不影響細胞存活(圖3C),表明ZBP1-RIPK3軸不能直接調節心肌細胞死亡。
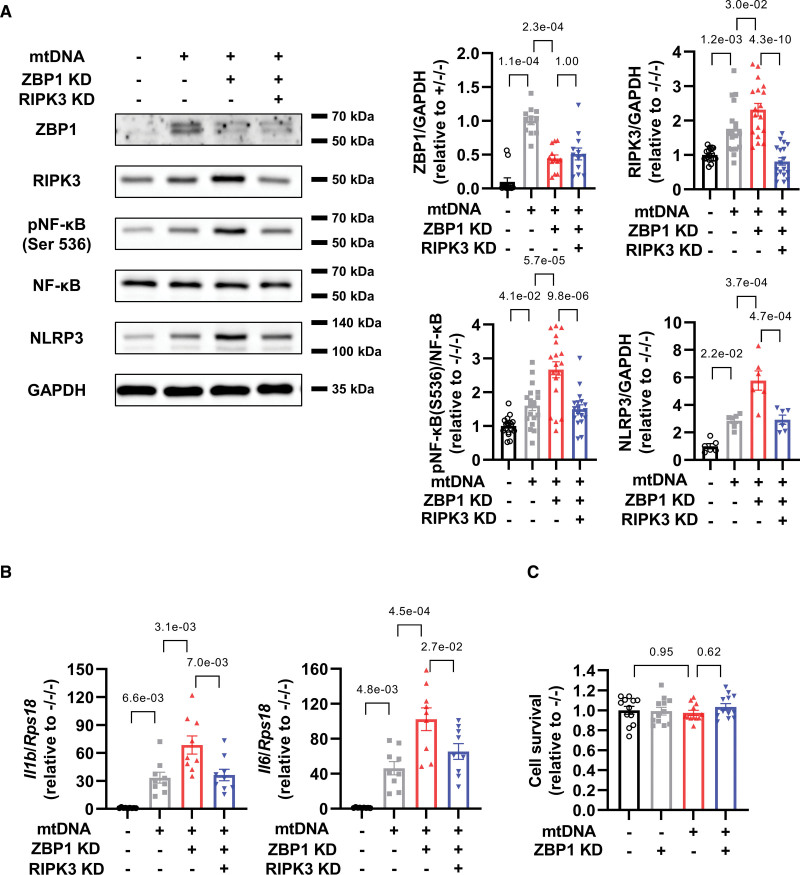
圖3在mtDNA處理的心肌細胞中,敲低RIPK3可通過敲低ZBP1抵消NF-κB-NLRP3軸的增加
4. 在mtDNA處理的心肌細胞中,敲低NF-κB抑制了ZBP1敲低引起的NLRP3軸的增加
接下來,作者研究了NF-κB是否可以介導炎癥作為ZBP1的下游靶標。作者發現NF-κB敲低減輕了mtDNA處理的心肌細胞中ZBP3敲低NLRP3增加的惡化,但不能減輕RIPK1的惡化(圖4A)。重要的是,這些變化伴隨著IL-1β和IL-6的mRNA水平的降低(圖4B)。綜上所述,這些數據表明ZBP1通過抑制NF-κB途徑發揮抗炎作用。
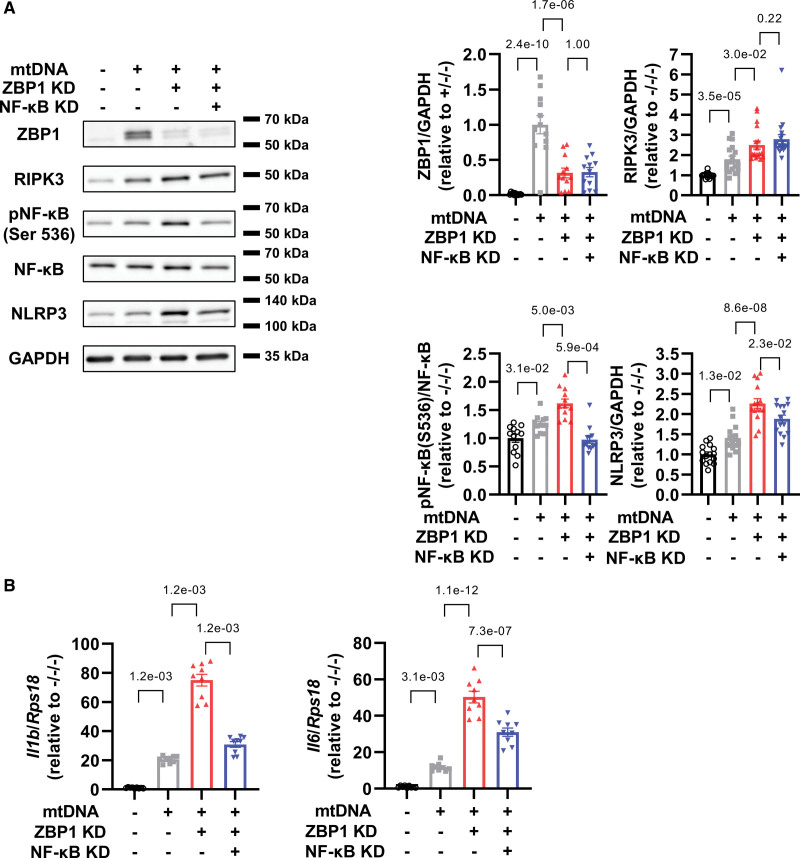
圖4 在mtDNA處理的心肌細胞中,敲低NF-κB抑制了ZBP1敲低引起的NLRP3軸的增加
5. ZBP1負調節CpG-寡脫氧核苷酸處理的心肌細胞中的炎癥反應
眾所周知,ZBP1可以感知線粒體和核DNA。為了鑒定刺激心肌細胞中ZBP1的DNA類型,作者使用了模擬mtDNA的CpG-寡脫氧核苷酸和模擬核DNA的對照寡脫氧核苷酸。作者發現異硫氰酸熒光素(FITC)標記的CpG-寡脫氧核苷酸與mcherry標記的ZBP1共定位(圖5A)。5CpG-寡脫氧核苷酸增加ZBP1 mRNA和蛋白質水平(圖5B和5C)和ZBP1敲低進一步增加了用CpG-寡脫氧核苷酸處理的心肌細胞中IL-1β和IL-6的mRNA水平(圖5D)。這些發現表明,ZBP1可以感知心肌細胞中的線粒體和核DNA,并負向調節炎癥以響應它們。
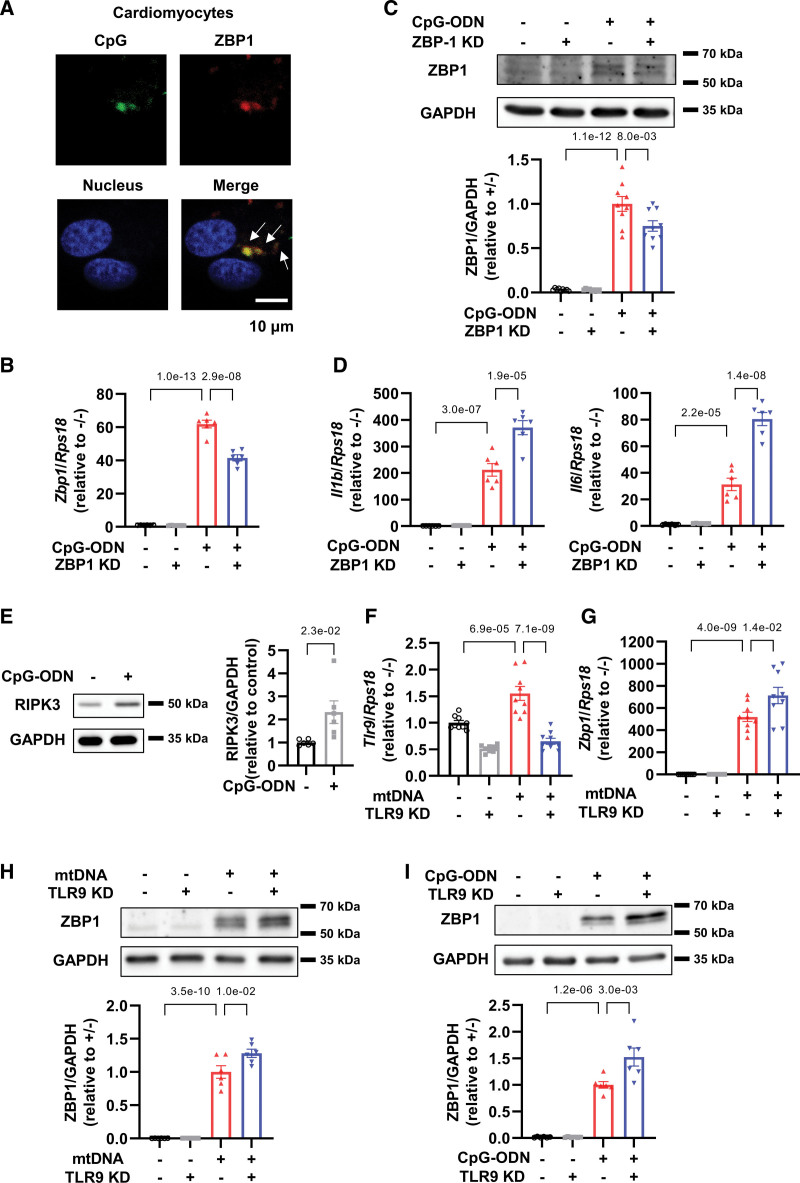
圖5 ZBP1和CpG-ODN的相互作用參與ZBP1在CpG-ODN處理的心肌細胞中的抗炎作用
6. TLR9 敲低通過抑制 RIPK3、NF-κB 和 NLRP3 來改善 mtDNA 或 CpG-寡脫氧核苷酸處理的心肌細胞中的炎癥反應
接下來,作者試圖研究mtDNA處理的心肌細胞中TLR9,ZBP1和RIPK3之間的關系。CpG-寡脫氧核苷酸,也稱為TLR9刺激劑,被發現增加心肌細胞中的RIPK3表達(圖5E),表明RIPK3是TLR9的下游。作者證實TLR9敲低有效地降低了其mRNA水平(圖5F)。此外,作者發現TLR9敲低增強了mtDNA誘導的心肌細胞中ZBP1 mRNA和蛋白質水平的增加(圖5G和5H)和加劇的CpG-寡脫氧核苷酸誘導的ZBP1增加(圖5I)。這些數據表明ZBP1阻礙了mtDNA和TLR9之間的相互作用,從而預防了下游炎癥信號傳導。
7. 心肌梗死后衰竭心臟的ZBP1和胞質mtDNA增加
為了評估ZBP1在衰竭心臟中的作用,作者在小鼠中進行了MI的體內實驗。作者在MI模型中驗證了梗死。作者發現,心肌梗死術后1天,心肌梗死后心臟非梗死區域的ZBP3蛋白水平升高(圖6A)。接下來,作者分析了MI模型中胞質部分的DNA水平。作者對GAPDH、線粒體標志物COX4和自噬標志物LC3進行了免疫印跡驗證了胞質組分的純度(圖6B)。Dloop是mtDNA的一種成分,在細胞溶質部分中在對照心臟中大量檢測到,并在MI后心臟中增加(圖6B)。相反,核DNA的組成部分Tert和B2m在對照和心肌梗死后心臟中均略有檢測到(圖6B)。總體而言,這些數據表明ZBP1與衰竭心臟中胞質mtDNA的增加有關。

圖6 ZBP1敲除通過 RIPK3-NF-κB-NLRP3途徑增強衰竭心臟的心肌炎癥
8. ZBP1 敲除加劇心功能不全和心肌梗死后重塑
ZBP1基因敲除小鼠在心肌梗死后左心室(LV)舒張和收縮壓直徑增加,左心室射血分數降低,心臟衰竭部分縮短(圖7A)。ZBP1敲除也加劇了全心重和左心室體重的增加(圖7B)。在病理分析中,ZBP1敲除增強了衰竭心臟的膠原蛋白體積,這是心臟纖維化的指標,但不是橫截面積,心肌細胞肥大的指數(圖7C)。這些數據表明,ZBP1 敲除會加劇心肌梗死后的心功能不全和重塑。
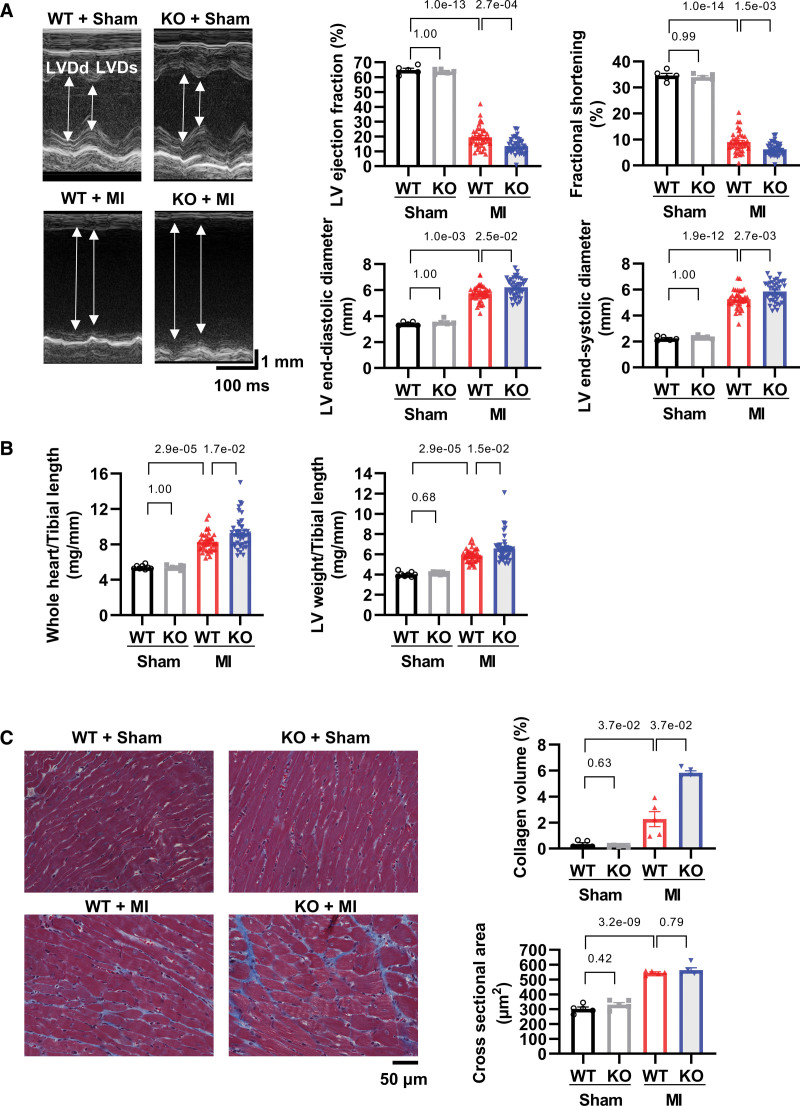
圖7 ZBP1敲除會加劇心肌梗死后的心功能障礙和重塑
結論:
ZBP1作為RIPK3-NF-κB通路的心肌炎癥內源性抑制因子,在心臟重塑中起保護作用。旨在通過ZBP1干擾心肌炎癥的治療策略可能有助于預防心臟重塑和衰竭。
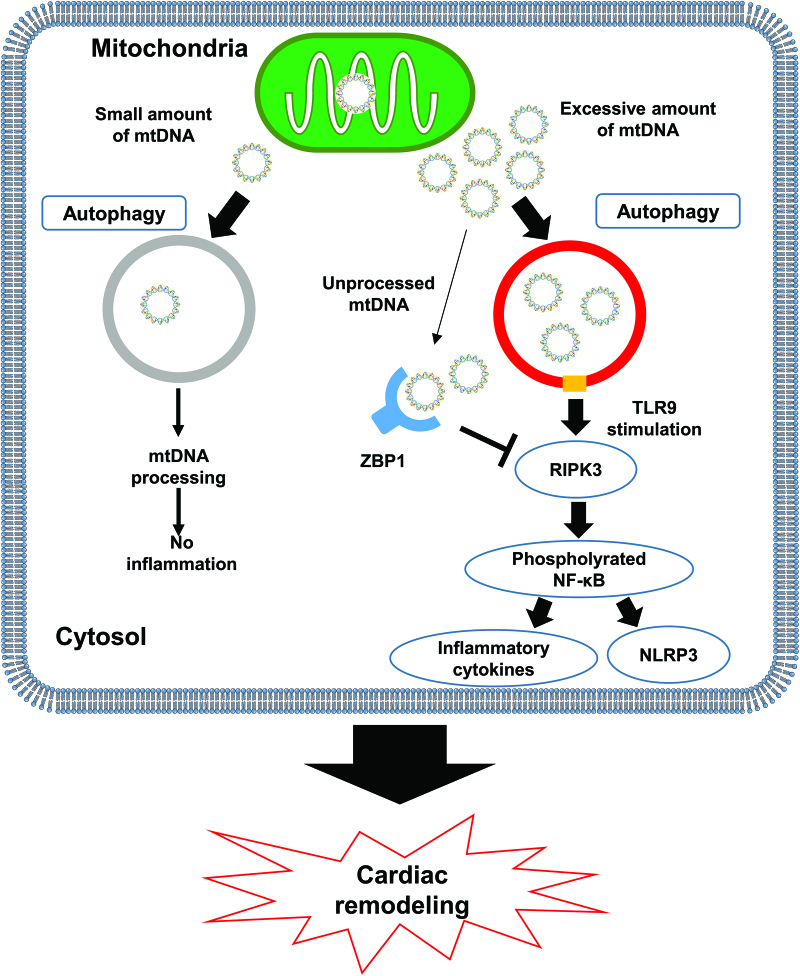
示意圖:
ZBP1在心肌炎癥和重塑中的作用示意圖
實驗方法:
細實時聚合酶鏈反應,免疫印跡,細胞活力檢測,動物實驗
參考文獻:
Enzan N, Matsushima S, Ikeda S, Okabe K, Ishikita A, Yamamoto T, et al. ZBP1 Protects Against mtDNA-Induced Myocardial Inflammation in Failing Hearts. Circ Res. 2023 Apr 28;132(9):1110-1126.