






你不懂的新機制——SRSF6調控可變剪接適應缺氧反應
缺氧誘導會導致可變剪接(AS)發生巨大變化,從而細胞適應缺氧,但是具體是何種剪切因子的AS幫助細胞適應缺氧的機制仍不清楚。SR蛋白是與可變外顯子結合并促進其納入成熟mRNA的重要剪接因子,其包括SRSF1至SRSF12在內的12個家族成員。SR蛋白在缺氧條件下以細胞類型特異性的方式被差異調節。然而,SR蛋白如何調控AS影響缺氧反應和癌癥進展的機制仍有較多未知之處。本研究發現SRSF4-PCE-SRSF6缺氧軸在不同腫瘤類型中均有活性,缺氧腫瘤中SRSF6高表達與不良預后相關。SRSF6的超保守PCE具有腫瘤抑制作用,并且PCE包含在缺氧中對降低SRSF6水平至關重要,這可能阻止腫瘤細胞進入缺氧適應的轉移途徑。本研究于2023年1月發表在《Nucleic Acids Research》IF:19.160期刊上。
技術路線:

主要實驗結果:
1、HeLa細胞通過整體外顯子跳變適應缺氧
為更好地理解AS介導的急性缺氧(24 h; 0.2% O2)適應機制,對HeLa細胞進行測序,并用MAJIQ分析AS。總計發現7178個局部剪接變異(LSVs)在缺氧后顯著(|ΔPSI|) >5%)改變(圖1A)。大多數LSV影響可變盒式外顯子(CEs)(4238,59%),其更常表現為跳躍性增加(圖1B)。通常,跳躍的外顯子比包含的外顯子更短(圖1C)。半定量RT-PCR驗證了9個AS事件,包括7個外顯子跳躍和2個外顯子包含事件(圖1D,E)。此外,還對RNA測序中的差異表達基因(DEGs)進行分析,GO發現RNA splicing是最顯著下調的生物學過程。其反映在SR家族蛋白(SRSF1, SRSF3, SRSF6, SRSF7 and SRSF8)的水平顯著下調。RT–qPCR證實SRSF3,SRSF6,SRSF7的表達在低氧組顯著下調(圖1F)。這些結果表明低氧降低剪接因子的表達并導致整體外顯子跳躍。
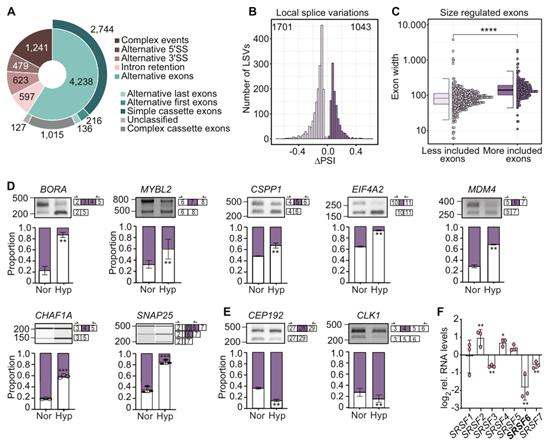
2、在急性缺氧條件下SRSF6水平和剪接活性顯著降低
在所有SR家族成員中SRSF6轉錄水平表現出最顯著下調,表明低水平SRSF6可能是缺氧條件下廣泛外顯子跳躍的部分原因。SRSF6的一致性保守域GAAGAA在缺氧跳躍外顯子中富集,而SRSF1的結合保守域GCA在被包含的外顯子中富集(圖2A)。與SRSF6 mRNA水平下調一致,缺氧24 h后SRSF6蛋白水平降低了60%;同時,p-SRSF6的水平顯著升高(圖2B)。為進一步研究缺氧對SRSF6亞細胞定位的影響,構建穩定低表達的GFP-tagged-SRSF6的海拉細胞系。結果顯示,在缺氧條件下,SRSF6–GFP也下調(圖2C,D,6A)。殘留的SRSF6-GFP僅在NSs中檢測到,其中磷酸化的SR蛋白通常以非活性形式儲存(圖2E)。在缺氧條件下,SRSF6的剪接功能通過轉錄物和蛋白質水平的降低以及磷酸化的增強而受損。
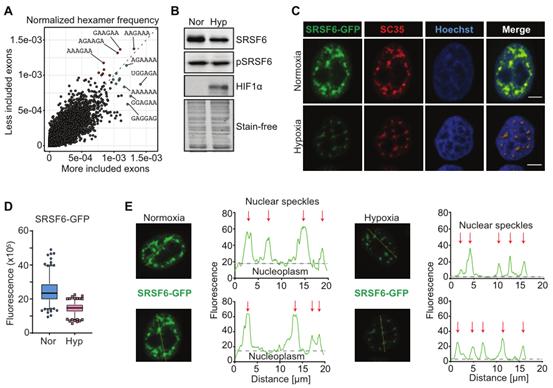
圖2在急性缺氧條件下,SRSF6水平和剪接活性顯著降低
3、缺氧條件下降低SRSF6的水平需要包含毒性盒式外顯子(PCE)結構域
不同癌癥類型中SRSF6表達增加與不良預后相關。因此,了解SRSF6活性在缺氧條件下被調節的機制可能具有治療意義。MAJIQ分析表明,在缺氧條件下,SRSF6的外顯子3的包含增加了約3倍(圖3A,B)。在常氧細胞中,CHX處理后,SRSF6-PCE和SRSF3-PCE的水平均顯著升高,從而抑制NMD(圖3B)。相比之下,兩種PCE亞型在缺氧狀態下被檢測到,NMD抑制沒有進一步的穩定作用(圖3B)。這表明NMD在缺氧狀態下會受損,PCE亞型不會通過這一途徑降解。
為探究PCE包含是否導致缺氧時的SRSF6降低,作用采用CRISPR/Cas9基因編輯法移除基因組SRSF6的3’剪接位點(SRSF6ΔPCE)。RT-qPCR揭示在三種PCE缺失的亞型中,SRSF的表達在缺氧時不再下降,都表現出增長趨勢(圖3D)。在缺氧條件下,PCE缺失克隆中不再包含PCE,重要的是,SRSF6蛋白水平沒有降低(圖3E)。總之,這些數據表明,SRSF6在缺氧中的減少需要包括PCE。

4、SRSF4通過PCE包含調控SRSF6水平
PCE包合通常由3’剪接位點下游30-60 nt的SR蛋白結合促進。iCLIP數據比較了常氧狀態下7種SR蛋白,顯示SRSF6之后,SRSF4是SRSF6 PCE的第二個最重要的結合劑(圖4A)。這表明SRSF6和SRSF4可能同時調節PCE包含和SRSF6在缺氧中的減少。為驗證這一點,生成并分類了SRSF4-GFP和SRSF6-GFP過表達的HeLa細胞(圖4B)。在常氧狀態下,SRSF6和SRSF4 OE均能降低內源性SRSF6蛋白水平(圖4B),它們在缺氧條件下進一步下降并伴隨著PCE包含的增加(圖3C,4C)。結合iCLIP數據,這提示,除了SRSF6自動調節外,缺氧狀態下SRSF4水平的增加通過包含SRSF6 PCE直接下調SRSF6水平(圖1F)。
為評估這一機制是否也存在于腫瘤中,分析了來自6個TCGA隊列的腫瘤樣本中的SRSF4和SRSF6表達水平以及PCE包含情況。值得注意的是,SRSF4和SRSF6水平在BRCA、COAD和STAD中呈顯著的反相關,除COAD外,SRSF4水平與PCE包含度呈正相關(圖4D)。作為對照,還分析了SRSF2和SRSF5,它們也與SRSF6 PCE結合,但頻率較低(圖4A)。納入BRCA、CESC和LIHC腫瘤樣本的缺氧評分證實,SRSF4水平與高缺氧評分呈正相關,而SRSF6水平與高缺氧評分呈負相關(圖4E)。總之,這些數據表明,在低氧性癌癥類型中,高SRSF4水平可能通過PCE包含降低SRSF6水平。
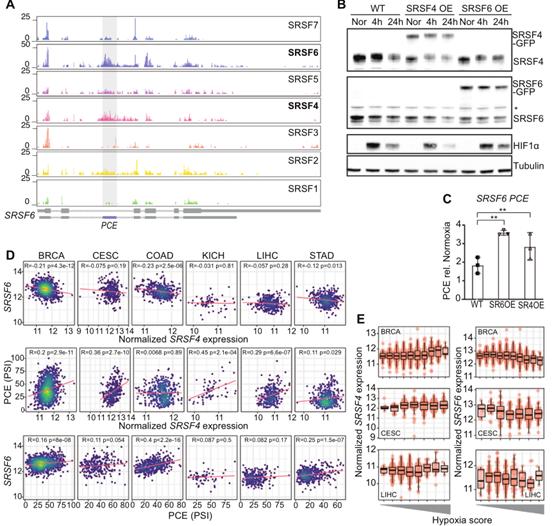
圖4 SRSF4調控SRSF6水平通過PCE包含
5、在缺氧狀態下維持高SRSF6水平會損害外顯子跳躍
為測試降低SRSF6水平是否對低氧適應很重要,研究了持續SRSF6過表達的影響。對SRSF6 OE細胞進行RNA-seq和MAJIQ分析,以確定常氧和24小時缺氧之間的剪接變化。總的來說,SRSF6 OE細胞在缺氧條件下比WT表現出更多的剪接變化(圖5A)。然而,與WT細胞相比,在SRSF6過表達細胞中,缺氧狀態下,受調節的盒式外顯子中的跳躍外顯子的比例明顯降低,表明外顯子跳躍可能受損(圖5B)。事實上,在WT細胞中鑒定出的1701個下調的LSVs中,1150個在SRSF6 OE細胞中沒有被調控,而有62個甚至被上調(圖5C-E)。
為評估SRSF6過表達是否會減弱缺氧條件下的外顯子跳躍和環化,從WT數據集中選擇了6個外顯子跳躍事件和3個缺氧誘導的circRNA。除CSPP1外顯子5外,所有被選中的跳躍或環狀外顯子均含有大量SRSF6結合基序(GAA)。RT-PCR證實這些外顯子在常氧下發生跳躍,在低氧下它們的跳躍增加,在所有情況下,在SRSF6 OE細胞中,它們的低氧驅動的跳躍減弱。引人注目的是,除CSPP1外顯子5外,這些外顯子的跳躍在ΔPCE細胞中也被減弱(圖5E)。總之,在缺氧狀態下,維持高SRSF6水平會損害外顯子跳躍。

6、缺氧條件下SRSF6與3’剪切位點的結合下降
為進一步研究SRSF6介導的AS在缺氧中的調節機制,進行iCLIP(圖6A)。選擇較早的4小時時間點來比較SRSF6與缺氧誘導轉錄本的結合,因為此時HIF1α水平最高,而SRSF6水平尚未降低(圖6A)。對匯集重復和嚴格篩選的交聯事件進行峰值調用,在3145個靶基因中鑒定出18956個可重復的SRSF6結合位點(圖6B)。共85.6%(16 234個)的結合位點定位于蛋白編碼基因,其中CDSs結合最為富集。富含嘌呤的共識結合基序(GAAGAA)在CDS區域富集(圖6C)。然而,在內含子序列中,SRSF6結合基序富集于尿苷,這表明SRSF6也可與多嘧啶束結合(圖6C)。SR蛋白通常通過結合外顯子剪接增強子(ESE)序列來促進剪接,但它們也可以通過結合內含子剪接沉默子(ISS)序列來抑制替代外顯子的包含。事實上,在SRSF6 OE細胞中,缺氧時,CEs 3 '剪接位點上游12-13 nt被跳過,內含子中可見SRSF6結合的明顯峰值(圖6D,紅色箭頭)。該內含子峰在更多的包含外顯子中不可見,且在24小時缺氧條件下顯著降低,而SRSF6與這些CEs兩側外顯子的結合沒有改變。在下游外顯子的3 '剪接位點可見到在缺氧時消失的類似峰值(圖6D,紅色箭頭)。SRSF6在跳躍外顯子中的結合率普遍高于包含外顯子,缺氧24 h后結合率下降(圖6D,藍色箭頭)。在SRSF6 OE細胞中專門調控的外顯子上也觀察到類似的缺氧敏感峰。總之,數據表明,SRSF6通過直接結合到可變外顯子或3 '剪接位點上游,促進常氧狀態下可變外顯子的包含。

7、缺氧時SRSF6下調導致核散斑擴散
除了在AS調節中起作用外,SRSF6也是NSs的核心蛋白。有趣的是,NSs在缺氧時擴散(圖7A,B)。使用標記物SC35和SRRM2進行免疫熒光實驗,在SRSF6水平較低的WT和SRSF4 OE細胞中,NSs在缺氧條件下看起來更小、更多或完全消失。然而,CoCl2處理后沒有觀察到NS擴散,SRSF6水平保持不變,并且SRSF6水平在SRSF6 OE或SRSF6ΔPCE細胞中也明顯減少,在缺氧條件下SRSF6水平仍然很高(圖7C,D)。這些數據表明,SRSF6在缺氧狀態下的減少是NS擴散所必需的。
文獻報道NS的擴散也會影響lncRNA MALAT1的流動性,它存在于這些凝析液中。根據iCLIP數據,MALAT1是SRSF6結合密度最高的轉錄本,在缺氧條件下進一步增加(圖7E,F)。SRSF6沿著MALAT1的整個長度密集包覆,支持SRSF6在將這個lncRNA拴在NS上的直接作用(圖7F)。當SRSF6水平較低時,MALAT1水平在缺氧時升高,lncRNA在NSs和核質中檢測到。相反,在SRSF6過表達細胞中,MALAT1僅在NSs中可檢測到,并與SRSF6和NS標記物SC35完全共定位(圖7G)。這說明在缺氧狀態下,SRSF6的下調和NS的擴散增加了MALAT1的核遷移率。為進一步測試這一點,作者量化了MALAT1的可萃取性,這與NSs或其他相分離的生物分子凝聚物中的隔離程度相關。在沒有熱處理的情況下,在缺氧的SRSF6 OE細胞中,MALAT1的可提取性明顯低于WT細胞,而WT細胞中MALAT1在沒有熱處理的情況下幾乎可以完全提取(圖7H)。數據表明,在缺氧條件下,高水平的SRSF6會阻止NS擴散和全局外顯子跳躍,并降低MALAT1的可萃取性和核遷移率。
為測試缺氧介導的SRSF6減少和NS擴散是否也發生在非癌細胞中,將HUVECs置于24小時的缺氧中。與HeLa細胞相似,SRSF6 mRNA水平在缺氧狀態下降低了約5倍(圖7I),SRSF6蛋白水平降低了約50%。Ss也在缺氧條件下分散(圖7J),MALAT1在此條件下完全可提取,相比之下,在常氧條件下,MALAT1的可萃取性減少了3.4倍(圖7K)。在測試的6個剪接靶點中,有3個顯示了與缺氧HeLa細胞相同的剪接模式(圖7L), MALAT1水平也增加了(圖7I)。這表明,由SRSF6減少介導的急性缺氧反應在HeLa細胞和HUVECs細胞中是保守的,但AS的結果可能是細胞類型特異性的,可能取決于轉錄組和SRSF6與其他剪接調節蛋白的相互作用。
總之,這些結果表明,在缺氧條件下,SRSF6的減少允許NS擴散,這導致了包括MALAT1在內的儲存的剪接因子的釋放。這促進了應激相關基因的外顯子跳躍和AS,并在低氧應激中提供了SRSF6介導的除直接影響AS外的另一層基因調控。
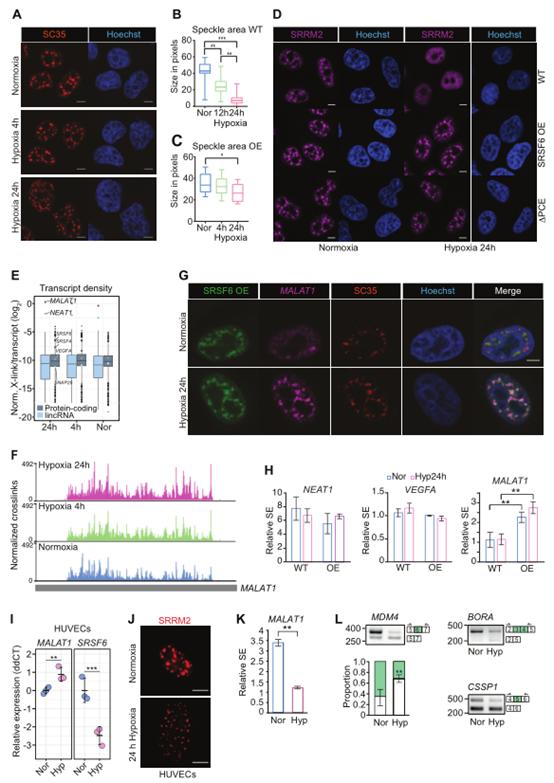
圖7 在低氧條件下,SRSF6的下調使NS擴散并增加MALAT1的核遷移率
8、在缺氧狀態下維持高水平的SRSF6抑制應激反應,并導致DNA損傷和基因組不穩定
為評估低氧條件下高SRSF6水平對細胞生存和適應的影響,進行了DEGs分析。與WT細胞相比,SRSF6過表達細胞在缺氧條件下顯示出更少的DEGs(582下調,1409上調),表明在SRSF6過表達細胞中缺氧反應被部分抑制。通過對不同條件下基因表達模式的聚類分析,發現了幾個聚類的DEGs,在這些DEGs中,在WT細胞中觀察到的缺氧誘導的上調或下調在SRSF6 OE細胞中減弱(圖8A)。在低氧條件下,SRSF6 OE細胞中HIF1α和HIF1α誘導的蛋白BNIP3的表達水平均低于WT細胞(圖8B)。在缺氧條件下,SRSF6 OE細胞的微核數量也明顯高于WT(圖8C,D),在DNA受損的細胞中,這是染色體不穩定的指標。SRSF6基因中的PCE失活引起了相同的染色體不穩定性表型(圖8C,D)。在WT細胞缺氧時清晰可見的應激顆粒,在SRSF6 OE和ΔPCE細胞中完全被抑制,這與高SRSF6水平抑制一般應激反應一致(圖8E)。

圖8 在缺氧條件下維持高水平SRSF6抑制壓力響應并導致基因組不穩定
總之,本研究確定SR蛋白SRSF6是控制缺氧時適應性AS反應的一個關鍵因素(圖9)。利用HeLa細胞發現,SRSF6的活性在急性缺氧時通過總蛋白水平的降低和SRSF6磷酸化的增強而降低。前者是通過以犧牲蛋白質編碼的SRSF6異構體為代價,將PCE納入其自身的轉錄本中實現的。PCE的包含主要是由SRSF4和SRSF6本身促進的,SRSF4的水平和活性在缺氧時增加,而SRSF6本身則與PCE大量結合。數據表明,SRSF6在缺氧時失活有雙重目的:(i)它允許SRSF6的直接目標外顯子跳躍和剪接特定的circRNA,這可能在缺氧適應中完成特定的功能;(ii)它在缺氧時引起NS的分散,釋放儲存的剪接因子,預計會引起基因表達和剪接的整體變化,正如在缺氧觀察到的那樣。
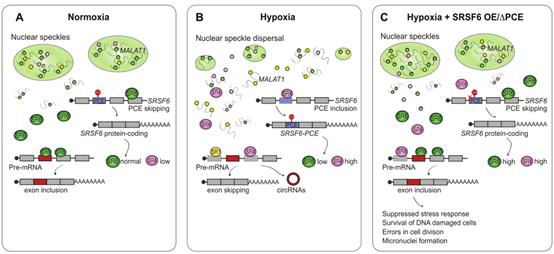
圖9 SRSF6調控缺氧適應的模式圖
參考文獻:
de Oliveira Freitas Machado Camila., Schafranek Michal., Brüggemann Mirko., Hernández Ca?ás María Clara., Keller Mario., Di Liddo Antonella., Brezski Andre., Blümel Nicole., Arnold Benjamin., Bremm Anja., Wittig Ilka., Jaé Nicolas., McNicoll Fran?ois., Dimmeler Stefanie., Zarnack Kathi., Müller-McNicoll Michaela.(2023). Poison cassette exon splicing of SRSF6 regulates nuclear speckle dispersal and the response to hypoxia. Nucleic Acids Res, 51(2), 870-890. doi:10.1093/nar/gkac1225