






PHB2通過對抗PKM2剪接維持VSMCs的收縮表型
血管平滑肌細胞(VSMCs)的表型轉變是多種血管疾病早期的發病機制。代謝重編程可能參與VSMC表型轉變。然而,將能量代謝與不同VSMC表型聯系起來的確切分子仍然難以捉摸。我們利用基因本體注釋收集細胞能量調節基因,并對轉化生長因子-β或血小板衍生生長因子BB刺激的VSMCs進行RNA-Seq分析。prohibitin2缺乏的VSMCs失去了可收縮的表型,這可以通過減少可收縮蛋白來證明。與Phb2flox/flox小鼠相比,Phb2SMCKO小鼠更容易損傷后VSMC增殖和新內膜形成。進一步的蛋白質相互作用組分析、免疫共沉淀和哺乳動物2-雜交實驗顯示,prohibitin 2通過其C端直接與丙酮酸激酶M1/2 (PKM) mRNA拼接的關鍵調節因子hnRNPA1相互作用,促進PKM2表達和糖酵解。Prohibitin2缺失促進PKM1/2 mRNA剪接和PKM1向PKM2的逆轉,并增強VSMCs的糖酵解。阻斷2-hnRNPA1相互作用導致PKM2表達增加,糖酵解增強,VSMCs中收縮標記基因表達抑制,以及體內損傷后新內膜形成加劇。總之,Prohibitin 2通過與hnRNPA1相互作用來抵消hnRNPA1介導的PKM選擇性剪接和葡萄糖代謝重編程來維持VSMC的收縮表型。本文于2022年10月發表于Circulation Research (IF=23.213)。
技術路線:

結果:
(1) RNA序列分析預測新的將能量穩態與VSMCs收縮表型聯系起來的候選基因
為了識別連接細胞能量代謝和VSMC表型的新候選基因,我們首先進行了RNA-seq分析,以揭示轉化生長因子-β (TGF-β)處理增強收縮表型相關基因表達或血小板衍生生長因子BB (PDGF-BB)處理降低收縮表型相關基因表達的原發性大鼠VSMC的轉錄組變化。在這1893個基因中,有6個基因與細胞能量調節基因數據庫重疊,包括prohibitin 2 (PHB2)、瞬時受體電位陽離子通道亞家族V成員4 (TRPV4)、ALK酪氨酸激酶受體(ALK)、組蛋白賴氨酸n-甲基轉移酶PRDM16(PRDM16)、乙酰輔酶a羧化酶2 (ACACB)和載脂蛋白C-III (APOC3)(圖1A)。接下來,我們驗證了mRNA水平,發現原發性大鼠VSMCs中PHB2、TRPV4和ALK的表達確實受到TGF-β和PDGF-BB的相互調節(圖1B)。接下來,我們使用靶向Phb2、Trpv4和Alk的特異性小干擾RNA (siRNAs)檢測VSMC收縮標志物(ACT A2、CNN1和SM22α)的mRNA水平。如圖1C所示,沉默Phb2、Trpv4和Alk可導致VSMC收縮標志物的表達顯著降低,提示這3個基因可能是連接VSMC能量代謝與收縮表型的候選基因。在這3個基因中,有報道稱prohibitin 2與prohibitin 1共同調控線粒體中的細胞色素-c氧化酶組裝和線粒體呼吸。然而,prohibitin 2在VSMC身份和血管穩態中的確切作用仍然未知。另外的免疫熒光分析顯示,prohibitin2主要定位于人動脈介質層(圖1D)。因此,我們探討了prohibitin 2是否調控VSMC表型。

圖1:RNA序列分析預測了連接血管平滑肌細胞(VSMC)能量穩態和收縮表型的新候選基因
(2) Prohibitin2減少與VSMCs轉分化相關
進一步的Western blotting分析顯示,在原發大鼠VSMCs中,PDGF-BB刺激后,prohibitin 2蛋白水平與VSMCs收縮標記物同時降低(圖2A)。相反,TGF-β刺激的VSMCs表現出更高水平的prohibitin 2和收縮標志物(圖2B)。血清饑餓是觸發VSMC表型從去分化狀態向分化狀態轉變的典型刺激。血清饑餓VSMCs中prohibitin 2水平升高(圖2C)。RT-qPCR分析證實,在人類VSMCs中也觀察到類似的改變(補充圖未展示)。值得注意的是,在VSMCs表型轉化過程中,prohibitin-1蛋白水平無統計學差異(圖2A-2C)。同樣,與假手術動脈相比,球囊損傷后3、7和14天,大鼠頸動脈中prohibitin 2的表達顯著降低,同時VSMCs收縮標記物減少,內膜面積增加(圖2D)。因此,western blotting分析、RT-qPCR和免疫熒光分析表明,與對照組乳腺內動脈相比,頸動脈內膜切除術患者動脈中prohibitin2的表達明顯降低(圖2E-2G)。
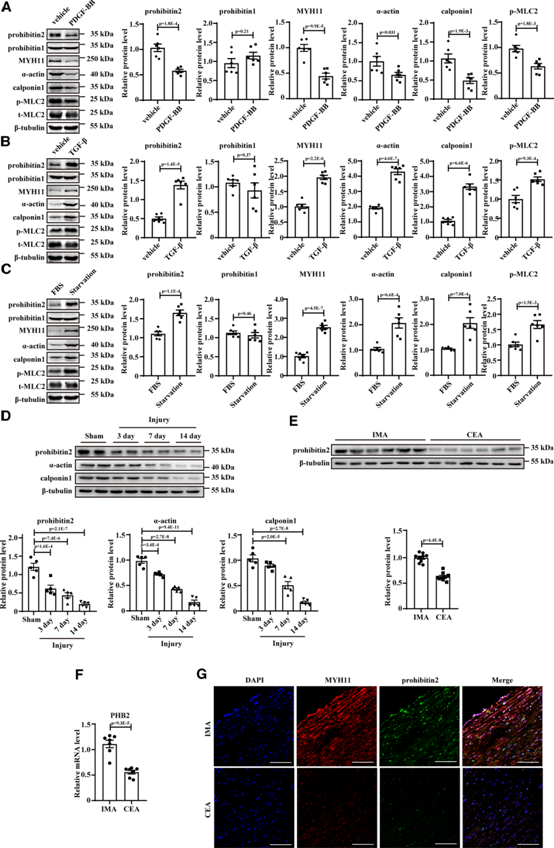
圖2:Prohibitin 2減少與血管平滑肌細胞(VSMC)去分化相關
(3) Prohibitin 2在體外維持VSMCs的收縮表型
我們探討了prohibitin2表達的干擾是否會影響VSMC表型切換。siRNA介導的人VSMCsprohibitin 2沉默可導致收縮標記物水平降低(補充圖未展示)。因此,TGF-β促進VSMCs收縮蛋白表達的上調通過prohibitin 2沉默被取消(圖3A)。為了進一步確定prohibitin 2在體內VSMC表型中的確切作用,將Phb2flox/flox小鼠與Tagln-Cre小鼠雜交,構建VSMC特異性Phb2缺失小鼠(Phb2SMCKO小鼠)(補充圖未展示)。與Phb2flox/flox小鼠(WTVSMCs)相比,Phb2SMCKO小鼠的原發性VSMCs (Phb2 KO VSMCs)中的VSMCs收縮標記物的mRNA和蛋白質水平明顯降低(圖3B)。F-actin染色顯示,prohibitin 2沉默導致原發大鼠VSMCs從細長收縮形態轉變為多邊形合成形態(圖3C)。此外,經Phb2siRNA轉染的VSMCs與經siRNA打亂轉染的VSMCs相比,收縮能力降低,膠原凝膠收縮試驗證明了這一點(siRNA打亂與siRNAPhb2: 46.3±3.86% vs 71.2±4.35%,P<0.05)(圖3D)。從Phb2SMCKO小鼠中分離出的主動脈環對苯腎上腺素的反應表現出收縮力減弱(圖3E)。在siRNAPhb2轉染的VSMCs中,細胞遷移活性也增加了,劃傷實驗和Boyden室實驗證明了這一點(圖3F和3G)。相反,prohibitin 2過表達2可挽救PDGF-BB誘導的VSMC收縮標記物減少和VSMC遷移(補充圖未展示)。綜上所述,這些數據表明禁用2是維持VSMCs收縮表型的關鍵。

圖3:Prohibitin 2在體外維持血管平滑肌細胞(VSMCs)的收縮表型
(4) VSMCs中Prohibitin 2的缺乏加劇體內損傷后新生內膜形成
對12周大的雄性Phb2SMCKO小鼠和Phb2flox/flox小鼠進行頸動脈鋼絲損傷。頸動脈MYH11 (VSMC標記物)和CD31 (EC標記物)免疫染色顯示,術后第0天,Phb2SMCKO小鼠和Phb2flox/flox小鼠的鋼絲損傷程度相同(補充圖未展示)。與Phb2flox/flox動脈相比,鋼絲損傷后第3、5和7天,Phb2SMCKO動脈中VSMC收縮標記物的表達減少,盡管新內膜形成沒有那么明顯的誘導(圖4A)。相反,損傷后14天,通過BrdU染色觀察到Phb2SMCKO動脈細胞增殖活性增強(圖4B)。結果,Phb2SMCKO小鼠的新內膜明顯大于Phb2SMCKO小鼠(圖4C)。此外,Phb2SMCKO動脈新內膜與中膜的比例遠高于Phb2flox/flox動脈,而中膜面積和外彈性層周長在兩組間無統計學差異。此外,為了排除taglncre介導的Phb2敲除可能引起的胚胎發育影響,我們將Phb2floxflox小鼠與Myh11-CreERT2小鼠雜交(補充圖未展示)。然后,8周齡雄性Phb2flox/flox小鼠和Phb2flox/floxMyh11-CreERT2小鼠用他莫西芬誘導出生后VSMCs的抑制性2耗盡。兩組小鼠(Phb2flox/flox+他莫西芬和Phb2flox/floxMyh11-CreERT+他莫西芬)頸動脈鋼絲損傷。與他莫昔芬處理的Phb2flox/floxMyh11-CreERT2小鼠相比,他莫昔芬處理的Phb2flox/flox/flox小鼠線損傷誘導的新內膜形成顯著增加(圖4D)。因此,prohibitin 2在體內維持VSMCs的收縮表型。
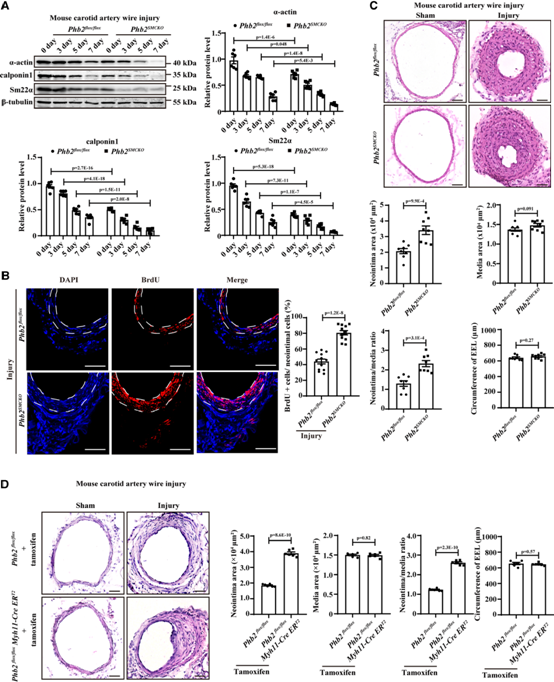
圖4:體內血管平滑肌細胞(VSMCs)的Prohibitin 2缺乏加劇損傷后新內膜的形成
(5) HnRNPA1是一種新的Prohibitin2結合蛋白
我們使用原發大鼠VSMCs來探索連接prohibitin 2和VSMCs表型調制的潛在結合伙伴。3個蛋白條帶被抗prohibitin 2抗體特異性免疫沉淀,而不是對照IgG。14個被抗prohibitin 2抗體免疫沉淀而非對照IgG免疫沉淀的蛋白被鑒定為潛在的prohibitin 2結合伙伴,包括prohibitin1 (PHB1)、異質核核糖核蛋白A1 (hnRNPA1)、顆粒蛋白(GRN)、PRMT1蛋白染色質靶蛋白(CHTOP)、14-3-3蛋白zeta (YWHAZ)、srsf1、膜聯蛋白A2 (ANXA2)等(圖5A)。我們首先分析了prohibitin2缺乏是否會導致hnRNPA1表達的改變。在鋼絲損傷小鼠頸動脈中,hnRNPA1的表達減少。因此prohibitin 2對hnRNPA1的表達沒有調控作用。接下來我們評估了prohibitin 2和hnRNPA1的潛在相互作用。在原代大鼠VSMCs的細胞裂解液中,內源性prohibitin 2與抗hnRNPA1抗體共免疫沉淀,但不與對照IgG共免疫沉淀,反之亦然(圖5B)。進一步的共免疫沉淀分析顯示,prohibitin 2與抗hnRNPA1抗體共免疫沉淀,但與對照IgG未在原代大鼠VSMCs的核部分共免疫沉淀,但在細胞質部分不共免疫沉淀,這表明prohibitin 2-hnRNPA1相互作用發生在細胞核(圖5C)。hnRNPA1和prohibitin2一致地共定位于原發性大鼠VSMCs的細胞核(圖5D)。HEK293A細胞分別轉染hnRNPA1 FL/ΔM9/ΔRGGbox-M9質粒旁的pcDNA3.1-6×his-prohibitin 2質粒和flag-CMV質粒(圖5E),然后使用抗flag抗體進行共免疫沉淀分析。hnRNPA1 FL和hnRNPA1ΔM9突變體均通過標志抗體特異性免疫沉淀出prohibitin 2,而不是對照IgG(圖5F)。hnRNPA1的RGG-box結構域與prohibitin 2結合(圖5G)。接下來,我們亞克隆了prohibitin 2的不同結構域(圖5H)。Flag-hnRNPA1通過抗flag抗體特異性免疫沉淀prohibitin 2的C端,但不通過對照IgG,反之亦然(圖5I)。通過哺乳動物2-雜交實驗驗證了prohibitin 2與hnRNPA1的C端相互作用(圖5J)。總之,通過其C端,prohibitin 2直接與hnRNPA1的RGG-box結構域相互作用。
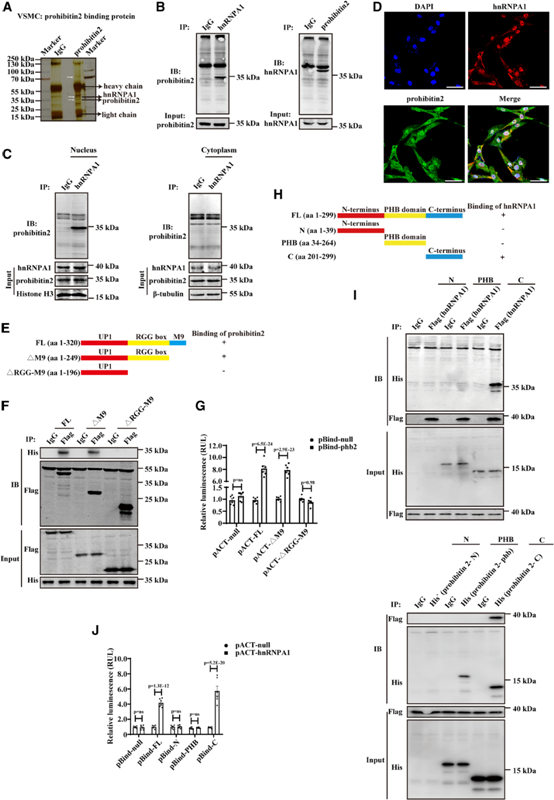
圖5:異質核核糖核蛋白A1 (HnRNPA1)是一種新型的prohibitin 2結合蛋白
(6) HnRNPA1-Prohibitin 2相互作用對維持VSMC體內外收縮表型至關重要
我們研究核內prohibitin 2和hnRNPA1的相互作用是否對VSMC的收縮表型至關重要。將帶有C端M9結構域的hnRNPA1的RGG-box結構域亞克隆到腺病毒pAdT機架-CMV載體中,重組腺病毒(Ad-Flag-RGG-M9)用于阻斷hnRNPA1和prohibitin2的核相互作用。免疫共沉淀實驗顯示,Ad-Flag-RGG-M9感染顯著阻斷了原發大鼠VSMCs中prohibitin 2-hnRNPA1相互作用(圖6A)。Ad-FlagRGG-M9感染抑制了α-actin和SM22α的轉錄活性,從而導致VSMCs去分化和細胞遷移增加(圖6B-6E)。Ad-Flag-RGG-M9感染加劇了球囊損傷后第7天VSMC收縮標記物的減少和新內膜形成的誘導(圖6F)。Ad-Flag-RGG-M9感染加重了大鼠球囊損傷模型28天新生內膜的形成(圖6G),而不會導致體重或血壓等顯著變化。免疫染色實驗證明,與假手術Ad-GFP感染大鼠頸動脈相比,球囊損傷Ad-GFP感染大鼠頸動脈VSMCs中prohibitin 2水平顯著降低(補充圖未展示)。在Ad-Flag-RGG-M9感染的大鼠頸動脈中,與Ad-GFP感染的大鼠頸動脈相比,prohibitin 2的水平沒有改變,這表明prohibitin 2-hnRNPA1相互作用不可能調控prohibitin 2的表達(補充圖未展示)。綜上所述,這些數據揭示了prohibitin 2-hnRNPA1相互作用對VSMC收縮表型維持的重要作用。
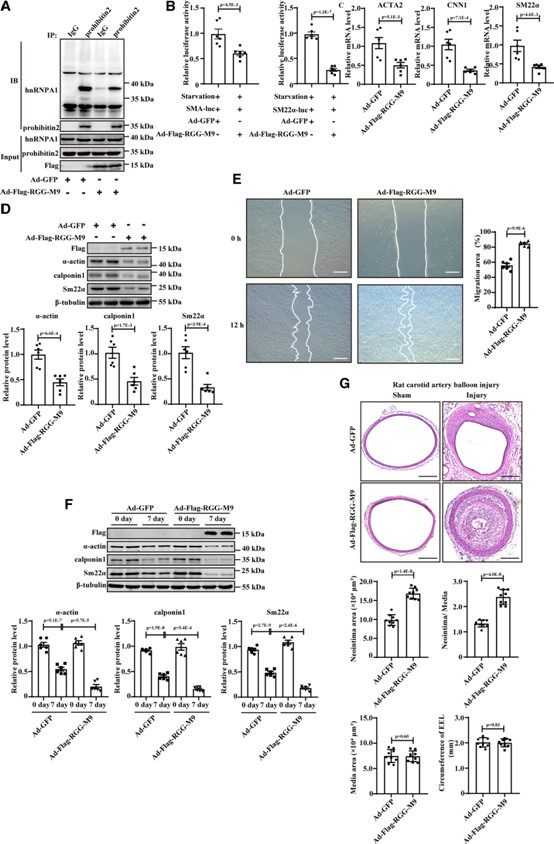
圖6:HnRNPA1-prohibitin 2相互作用對于維持血管平滑肌細胞(VSMCs)在體內外的收縮表型至關重要
(7) Prohibitin 2抑制hnRNPA1介導的pre-PKM mRNA剪接、PKM2表達和有氧糖酵解
hnRNPA1是pre-PKM mRNA剪接的主調控因子。PKM2是PKM的首選剪接異構體,調節糖酵解的最終限速步驟,而PKM1促進OXPHOS。這兩種異構體來自于PKM前mRNA的互排性選擇性剪接,反映了外顯子9 (PKM1)或外顯子10 (PKM2)的包含。hnRNPA1抑制外顯子9的序列側翼,導致外顯子10的包含和PKM2的表達。我們研究prohibitin 2是否影響hnRNPA1介導的pre-PKM mRNA剪接、PKM2的產生和有氧糖酵解。與Phb2flox/flox小鼠(WTVSMCs)相比,從Phb2SMCKO小鼠分離的原發性小鼠VSMCs (Phb2 KO VSMCs)中PKM2的mRNA水平升高,而PKM1的mRNA水平下降(補充圖未展示)。相比之下,pre-PKM mRNA水平沒有顯著改變,表明存在另一種剪接。PstI和NcoI酶切擴增子后進一步RTPCR顯示Phb2缺失促進PKM1向PKM2的反式表達(圖7A)。在Phb2 KO原發小鼠VSMCs(圖7B)、Ad-Flag-RGG-M9感染的大鼠VSMCs(圖7C)以及感染Ad-Flag-RGG-M9的球囊損傷大鼠頸動脈中使用western blotting也觀察到了類似的結果。Phb2 KO VSMCs糖酵解活性增強,OXPHOS活性降低,細胞糖酵解和有絲分裂應激試驗證明(圖7D)。PDGF-BB刺激的原發大鼠VSMCs中prohibitin 2被抑制的轉分化通過PKM2過表達被取消(圖7E)。為了進一步闡明prohibitin2是否在體內通過抑制PKM2的表達來維持VSMC的收縮表型和對抗新生內膜的形成,我們使用帶有SM22α啟動子的AAV9載體選擇性下調VSMC中的PKM2。在Phb2SMCKO小鼠頸動脈鋼絲損傷后第7天,VSMC特異性PKM2耗損挽救了VSMC收縮標記物的減少(補充圖未展示)。Phb2SMCKO小鼠鋼絲損傷后血管收縮力下降和新生內膜形成增強均因VSMC特異性PKM2耗損而得到挽救(圖7F-7G)。綜上所述,prohibitin2與核hnRNPA1直接相互作用,抑制hnRNPA1介導的pre-PKM mRNA剪接和PKM2的產生,從而調節葡萄糖代謝重編程,在體外和體內維持VSMCs的收縮表型。
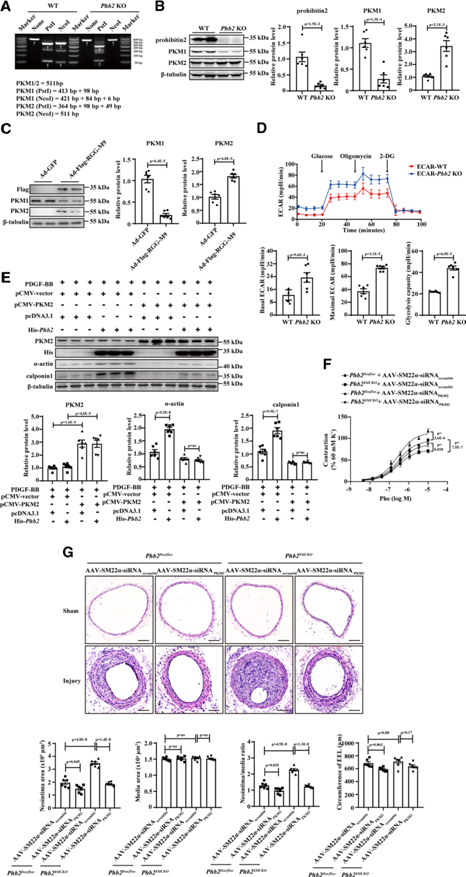
圖7:Prohibitin2抑制hnRNPA1介導的pre-PKM mRNA剪接、丙酮酸激酶異構體M2 (PKM2)表達和葡萄糖代謝重編程
結論:
Prohibitin2通過直接與核hnRNPA1相互作用,抑制hnRNPA1介導的pre-PKM mRNA剪接、PKM2的產生,從而調節葡萄糖代謝重編程(圖8),維持VSMCs的收縮表型,抑制損傷后新內膜的形成。Prohibitin2是維持VSMCs穩態的內源性糖酵解調節劑。以prohibitin2-hnRNPA1-PKM2軸為靶點調節VSMC能量代謝可能有助于心血管疾病的治療。
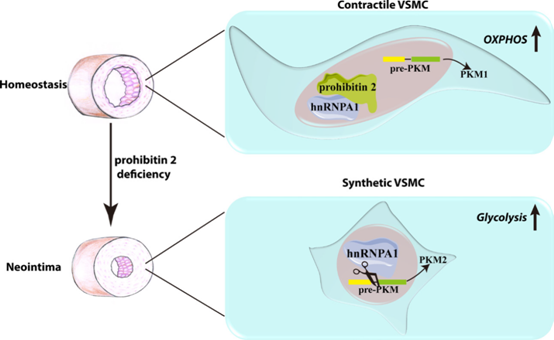
圖8:Prohibitin2在血管平滑肌細胞(VSMC)表型測定的示意圖
參考文獻:
Jia, Y., Mao, C., Ma, Z., Huang, J., Li, W., Ma, X., Zhang, S., Li, M., Yu, F., Sun, Y., Chen, J., Feng, J., Zhou, Y., Xu, Q., Zhao, L., Fu, Y., & Kong, W. (2022). PHB2 Maintains the Contractile Phenotype of VSMCs by Counteracting PKM2 Splicing. Circulation research, 131(10), 807–824. https://doi.org/10.1161/CIRCRESAHA.122.321005.