






凝血酶誘導腦缺血/再灌注過程中ACSL4依賴性鐵下垂
缺血性中風對人類,特別是老年人是一個重大的危險。干預措施只能用于清除血栓,缺血性中風期間神經(jīng)元死亡的機制仍存在爭議。鐵下垂越來越被認為是各種器官缺血后細胞死亡的一種機制。在這里,我們報道了絲氨酸蛋白酶,凝血酶,通過促進花生四烯酸的動員和隨后的鐵性基因,酰基輔酶a合成酶長鏈家族成員4 (ACSL4)的酯化來激發(fā)鐵性信號傳遞。一種無偏多組學方法確定了凝血酶和ACSL4基因/蛋白質(zhì),以及它們的前鐵毒磷脂酰乙醇胺脂質(zhì)產(chǎn)物,在嚙齒動物大腦中動脈閉塞時顯著改變。在基因或藥理學上抑制該途徑中的多個點,可減弱體外和體內(nèi)缺血模型的結(jié)果。因此,凝血酶-ACSL4軸可能是改善缺血性腦卒中鐵性神經(jīng)元損傷的關鍵治療靶點。本文于2022年2月發(fā)表于Signal Transduction and Targeted Therapy (IF=38.104)上。
技術(shù)路線:
主要研究結(jié)果:
(1) 急性腦I/R后凝血酶上調(diào)
由鐵下垂引起的細胞死亡是由于有毒的脂質(zhì)氫過氧化物積累超過細胞解毒的能力。花生四烯酸(AA)含有磷脂酰乙醇胺(PEs),特別容易發(fā)生鐵質(zhì)過氧化反應。通過對小鼠缺血性中風后海馬組織進行高質(zhì)量的非靶向代謝組學分析(圖1a),我們發(fā)現(xiàn)了四種磷脂顯著降低,即PC(36:4)、PE(42:8)、PE (38: 6p)和PE(40:7)(圖1b-h)。進一步分析其脂肪酸側(cè)鏈,發(fā)現(xiàn)PC(36: 4,16: 0/ 20:4)、PE(42: 8,22:4 / 20:4)、PE (38: 6p, 18: 2p/ 20:4)脂肪酸側(cè)鏈中均含有AA(圖1i),且AA(20:4)占主導地位(圖1j),這與小鼠缺血性腦卒中后海馬組織游離AA顯著增加(腦卒中/對照比= 1.49,p = 0.0007, Student’s t檢驗,圖1k)一致。
為了探索缺血性卒中后AA升高的蛋白,我們對同一組織進行了蛋白質(zhì)組學分析(圖2a-c)。基于差異表達蛋白的富集,我們發(fā)現(xiàn)血小板激活、信號轉(zhuǎn)導和聚集是上調(diào)最多的通路(圖2d,e)。蛋白質(zhì)-蛋白質(zhì)相互作用(PPI)網(wǎng)絡分析也一致指出了相同的途徑(圖2f),凝血酶原(F2基因編碼凝血酶原,也稱為凝血因子II),凝血酶原酶是上調(diào)最多的蛋白質(zhì)(stroke/control ratio = 10.88, p < 0.0001, Student 's t檢驗,圖2g),Western Blotting進一步證實了這一點(圖2h)。
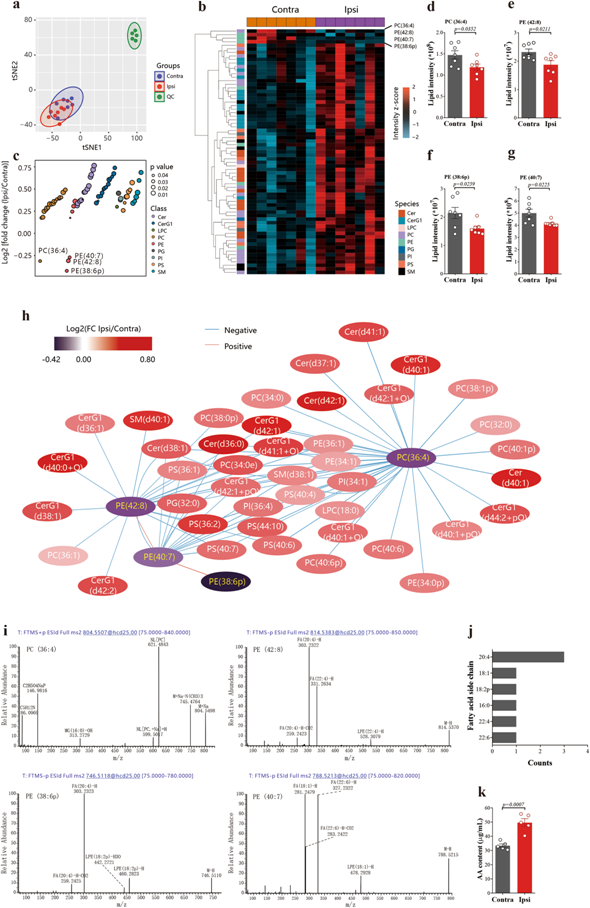
圖1:在缺血/再灌注后,磷脂側(cè)鏈上的花生四烯酸被釋放
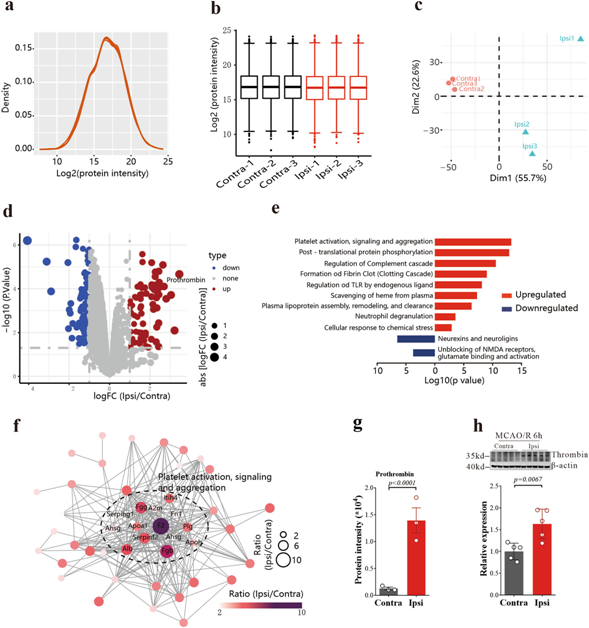
圖2:急性腦缺血/再灌注后凝血酶上調(diào)
(2) 凝血酶誘導鐵下垂獨立于鐵積累
凝血酶是缺血性腦卒中的主要藥物靶點之一,其作用是促進纖維蛋白的產(chǎn)生和凝血。我們發(fā)現(xiàn)凝血酶劑量依賴性誘導N27神經(jīng)元細胞死亡(圖3a),并伴有AA升高(圖3b)和脂質(zhì)氫過氧化物升高(3c,d),線粒體變小,膜密度增加(圖3e),這些特征都與鐵下垂一致。凝血酶不影響細胞內(nèi)亞鐵的水平,這不是誘導鐵下垂的必要條件(圖3f)。凝血酶誘導的細胞死亡可以通過鐵下垂抑制劑liproxstatin-1(圖3g)和谷胱甘肽前體n-乙酰-l-半胱氨酸(圖3h)和cPLA2α抑制劑darapladib(圖3i)的治療來阻止。利prox他汀-1同樣可以防止MDA-MB-231細胞(一種人類乳腺癌細胞系)的細胞死亡,這表明凝血酶在腦外鐵下垂中的作用(圖3j,k)。
凝血酶升高是缺血性中風后AA動員的上游信號,這有助于鐵下垂。達比加群在N27神經(jīng)元培養(yǎng)的I/R氧-葡萄糖剝奪(OGD)模型中保護細胞,神經(jīng)癥狀緩解和減少腦梗死體積(補充圖未展示)。由于OGD和MCAO模型都不涉及可能與凝血酶抑制有關的血栓形成,凝血酶誘導的另一種損傷被達比加群阻斷。這些數(shù)據(jù)強調(diào)了凝血酶誘導鐵下垂的可能性,這可能與缺血性中風有關。
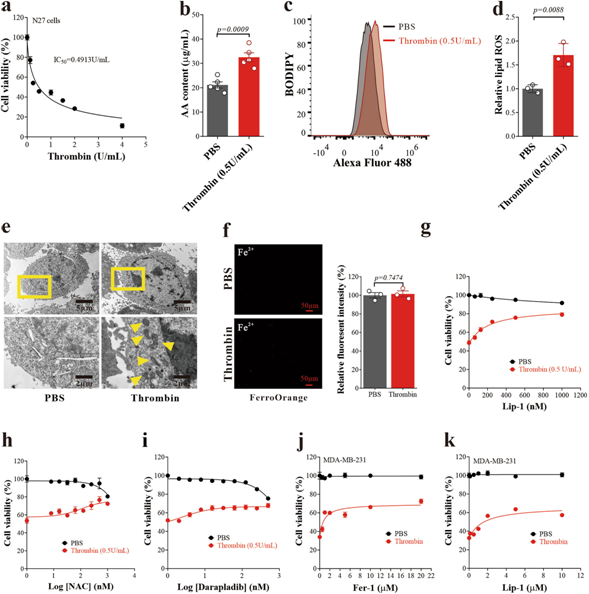
圖3:凝血酶誘導神經(jīng)元鐵下垂
(3) 腦I/R損傷發(fā)生氧依賴性鐵下垂
為了證實缺血性腦卒中中鐵墜癥的發(fā)生,并剖析引發(fā)鐵墜癥的必要條件,我們調(diào)節(jié)了MCAO/R的條件,并研究了其生化和行為變化。小鼠和大鼠在MCAO/R后(再灌注后24小時),與對側(cè)“對照”半球相比,同側(cè)“卒中”半球受影響的海馬和皮層區(qū)域的脂質(zhì)過氧化物(LPO)和丙二醛(MDA)(脂質(zhì)過氧化物的分解產(chǎn)物)水平顯著升高(小鼠:圖4a,b;大鼠:補充圖未展示)。透射電鏡(TEM)圖像顯示,MCAO/R后24 h同側(cè)皮層區(qū)域的這些特征伴有神經(jīng)元線粒體收縮,鐵脫鐵特征(補充圖未展示)。
在MCAO/R后立即給藥的亞毒性劑量(沒有明顯的神經(jīng)元損失)RSL3 (GPx4抑制劑)和erastin (Xc抑制劑)增加鐵毒傾向,顯著加重小鼠腦I/R損傷,表現(xiàn)為MDA水平顯著升高(圖4c),神經(jīng)行為缺陷(圖4d),梗死體積增加(MCAO/R后24小時;圖4e)。在MCAO小鼠模型中,脂基捕獲劑和原型鐵脫鐵抑制劑利普司他汀-1和鐵司他汀-1已被證明能有效地減弱腦I/R損傷。然而,這些抑制劑不能挽救永久性MCAO(即無再灌注)造成的神經(jīng)元損傷(圖4f,g),因為鐵上墜癥依賴于氧氣,進一步表明鐵上墜癥可能發(fā)生在再灌注階段。
我們在I/R的氧葡萄糖剝奪(OGD)細胞培養(yǎng)模型中證實了神經(jīng)元鐵下垂的發(fā)生。N27神經(jīng)元細胞經(jīng)OGD處理2小時(圖4h)死亡前,細胞內(nèi)Fe2+水平升高(圖4i),脂質(zhì)過氧化物(通過BODIPY 581/591 C11 [BODIPY]測定;圖4j,k),脂質(zhì)氫過氧化物(Liperfluo探針),以及細胞內(nèi)總ROS和MDA(補充圖未展示)。與對照細胞相比,OGD處理的N27細胞中的線粒體也顯著變小,但膜密度增加(補充圖未展示)。利prox他汀-1治療挽救了OGD誘導的神經(jīng)元死亡(圖1l),顯示了鐵下垂的參與。
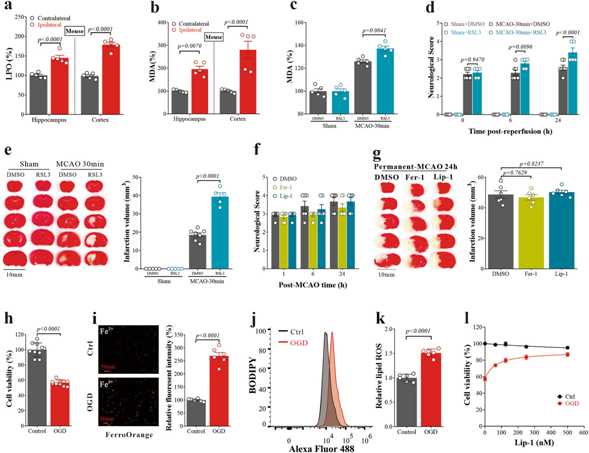
圖4:腦再灌注損傷的氧依賴性鐵下垂
(4) 缺血性卒中后血小板活化
凝血功能的激活是缺血性腦卒中中神經(jīng)元損傷的主要原因。神經(jīng)元中的凝血酶也可以通過其絲氨酸蛋白酶活性引發(fā)鐵下垂。為了研究缺血性腦卒中期間凝血系統(tǒng)的變化,我們收集了59份血清樣本,包括27例健康對照組和32例缺血性腦卒中患者,在去除血清中高豐度蛋白后,進行TMT標記的蛋白質(zhì)組學分析(圖5a-e)。84.7%的樣本相關值大于0.98,相關值范圍在0.968~0.993之間(圖5a),表明結(jié)果具有可重復性。在分析中,我們總共鑒定出584個蛋白質(zhì),其中424個蛋白質(zhì)可以得到≥2個多肽的支持,平均數(shù)量為8.31個(圖5c)。評估了不同樣本中蛋白質(zhì)的分布,我們發(fā)現(xiàn)超過50%的樣本中有362種蛋白質(zhì)被定量(圖5d)。基于包含362個蛋白質(zhì)的隨機森林估算數(shù)據(jù)的tSNE分析驗證了不存在批效應(圖5e)。
我們在缺血性卒中血清中共鑒定出28個蛋白上調(diào),21個蛋白下調(diào)(圖5f)。其中,缺血性腦卒中患者血清中纖維蛋白凝塊形成明顯上調(diào)(圖5g-k),這與小鼠大腦中血小板聚集增加一致(圖2f)。然而,與健康對照組相比,血清中凝血酶水平?jīng)]有明顯升高(圖5l),這表明我們在大腦中觀察到的升高(圖2h)可能不是來自血液。如果達比加群具有缺血性卒中后的神經(jīng)保護作用(補充圖未展示),其有益作用不太可能來自于其在血液中的抑制凝血活性。相反,缺血性中風患者血液中凝血酶沒有升高,這表明抗凝血酶藥物的任何益處都可能是由于大腦中凝血酶的升高(圖2h)。考慮到凝血酶在動員鐵墜體脂肪酸AA方面的額外作用,我們假設凝血酶可能會誘導缺血性中風后的鐵墜癥。

圖5:缺血性卒中后血小板活化
(5) ACSL4介導凝血酶神經(jīng)毒性
酰基輔酶a合成酶長鏈家族成員4 (ACSL4)是一種重要的脂質(zhì)代謝酶,通過將游離AA轉(zhuǎn)化為花生四烯醛-CoA11生成脂質(zhì)氫過氧化物來參與鐵脫鐵。與游離AA通過促進其泛素化和蛋白酶體降解抑制ACSL4水平一致,我們發(fā)現(xiàn)凝血酶處理后N27細胞中的ACSL4降低(圖6a),而不影響ACSL4 mRNA的表達。I/R后6 h,缺血區(qū)海馬區(qū)ACSL4蛋白表達明顯下降,而ACSL4 mRNA表達在同一時間點無明顯變化(補充圖未展示)。由于缺血性卒中后小鼠海馬區(qū)游離AA顯著增加(圖1k),在I/R過程中ACSL4的降低可能是翻譯后修飾的結(jié)果。通過Western Blot(圖6b)或組織學(圖6c)檢測,MCAO/R后大鼠同側(cè)海馬ACSL4的表達隨著時間的推移相似地下降,在I/R后3小時,ACLS4明顯減少,這是在I/R后6小時開始海馬神經(jīng)元損失(使用NeuN評估存活神經(jīng)元,使用Fluoro-Jade評估退化神經(jīng)元)之前(圖6d,e)。大鼠的結(jié)果與MCAO后小鼠的結(jié)果一致。然而,凝血酶在缺血早期明顯升高(圖6f)。這些數(shù)據(jù)表明,ACSL4下調(diào)是腦I/R的早期事件,獨立于神經(jīng)元死亡而發(fā)生,這可能是對凝血酶誘導應激的保護性反應。
為了闡明凝血酶細胞毒性是否由ACSL4介導,我們使用基于CRISPR-Cas9的基因編輯生成了ACSL4敲除(KO) N27細胞,使用含有ACSL4表達框的慢病毒載體mach-OE-rACSL4生成了ACSL4過表達(OE)細胞。Western Blotting證實了這兩種調(diào)制(圖6g,上圖)。凝血酶依賴于ACSL4引起毒性,因為凝血酶的細胞毒性因ACSL4的還原而恢復,并因ACSL4的過表達而加重(圖6g)。ACSL4抑制劑吡格列酮(PIO)也能阻斷凝血酶的細胞毒性(補充圖未展示)。這些結(jié)果表明,凝血酶可能通過促進ACSL4依賴的鐵墜瘤而導致神經(jīng)元細胞死亡,而ACSL4的降低可能有助于抑制凝血酶引起的鐵墜瘤損傷。

圖6:凝血酶誘導急性腦缺血/再灌注后ACSL4下調(diào)
(6) 調(diào)節(jié)ACSL4表達改變急性缺血性腦損傷的結(jié)局
為了確定ACSL4是否影響大腦I/R后的結(jié)果,我們使用單次注射腺相關病毒載體,即AAV8-mACSL4和AAV8-EF-Cas9+AAV8-mACSL4-sp,在小鼠左海馬CA3區(qū)域(小鼠MCAO/R最脆弱的區(qū)域)選擇性過表達或敲除ACSL4。并檢查了MCAO嚙齒動物模型的影響(圖7a)。以AAV8-EGFP(綠色熒光蛋白增強型AAV8)為對照。免疫熒光染色證實了轉(zhuǎn)導的有效性(圖7b)。在相同的30分鐘MCAO/R條件下,ACSL4 OE小鼠表現(xiàn)出明顯的梗死體積增加(灌注后24小時,圖7c),神經(jīng)評分惡化(圖7d),旋轉(zhuǎn)運動性能變差(圖7e)。與EGFP小鼠相比,ACSL4 KO小鼠對I/R損傷有保護作用,梗死體積減小(圖7f),神經(jīng)評分(圖7g)和運動協(xié)調(diào)性(圖7h)也有顯著改善。與鐵脫落抑制劑一致,ACSL4 KO小鼠永久性MCAO后神經(jīng)元損傷與對照小鼠無差異(圖7i,j),這強烈提示在該模型中,鐵脫落發(fā)生在再灌注期間。
激光散斑信號顯示敲除ACSL4后不影響MCAO/R后大鼠皮層的血流(圖7k,l),證實ACSL4 KO不是單純通過影響血流動力學而起到保護作用。AAV輔助ACSL4敲除大鼠皮層對I/R誘導的功能損傷具有顯著保護作用(神經(jīng)評分;圖7m)和腦梗死體積(TTC染色;圖7n)。
ACSL4 KO細胞對RSL3誘導的鐵墜病有保護作用,而ACSL4 OE細胞對同樣的毒素脆弱(補充圖未展示)。將這些細胞進行OGD/再氧化,發(fā)現(xiàn)ACSL4 KO細胞對OGD相關毒性具有抗性,而ACSL4 OE細胞更容易受到OGD相關毒性的影響(補充圖未展示)。
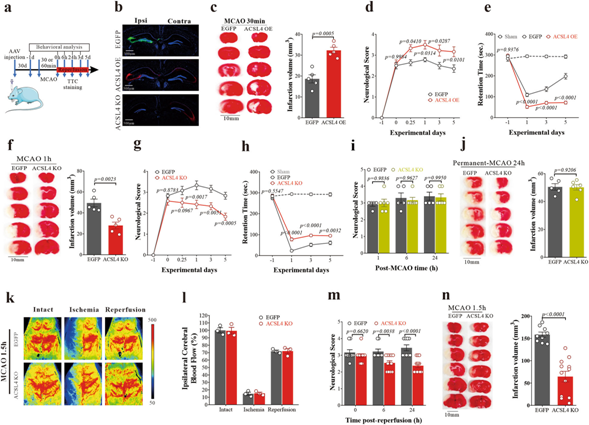
圖7:調(diào)節(jié)ACSL4表達改變急性缺血性腦損傷的結(jié)局
結(jié)論:總之,我們的研究結(jié)果有力地證明,抗凝血酶療法可能通過抑制鐵下垂而對腦卒中再灌注后有益,也可能對涉及鐵下垂的其他疾病有用。
參考文獻:
Tuo, Q. Z., Liu, Y., Xiang, Z., Yan, H. F., Zou, T., Shu, Y., Ding, X. L., Zou, J. J., Xu, S., Tang, F., Gong, Y. Q., Li, X. L., Guo, Y. J., Zheng, Z. Y., Deng, A. P., Yang, Z. Z., Li, W. J., Zhang, S. T., Ayton, S., Bush, A. I., … Lei, P. (2022). Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal transduction and targeted therapy, 7(1), 59. https://doi.org/10.1038/s41392-022-00917-z.