






樹突狀細胞功能微調
樹突狀細胞(DC)微調炎癥和耐受性反應以保護免疫病理。然而,共同調節因子在維持這種平衡方面的作用尚未得到探討。本研究發現NCoR1及其同源物NCoR2(SMRT)可通過調節STAT3信號通路維持DC炎癥和耐受性反應平衡。本研究于2022年9月發表在《Frontiers in Immunology》IF:8.786期刊上。
技術路線:

主要實驗結果:
1、SMRT KD cDC1 DCs增強激活/共刺激
構建穩定的Ncor2基因敲除(SMRT KD)和空載體對照的cDC1細胞系。敲除效率在mRNA水平和蛋白水平上得到驗證(圖1A)。然后,用RT-qPCR檢測對照組和SMRT KD cDC1細胞在CpG激活前后2小時和6小時促炎因子(Il12b)和抗炎因子(Il10)mRNA的表達。發現,與對照細胞相比,在SMRT KD cDC1中,CpG刺激后Il12b顯著增加,Il10表達同時減少(圖1A)。此外,流式細胞術分析顯示,與對照組相比,SMRT KD cDC1中的陽性細胞比例和共刺激分子(CD80、CD86和CD40)顯著增加(圖1B)。接下來,通過MHC-II和MHC-I在SMRT KD cDC1中的表達來研究抗原呈遞能力。在CpG和pIC激活過程中,MHC-I陽性細胞比例顯著增加,而MHC-II水平保持不變(圖1)。MHC-II和MHC-I分子的MFI變化也表現出類似的趨勢。在CpG刺激18h后,用流式細胞術檢測CD80、CD86和MHC-II的表達,發現SMRT KD BMcDC1較對照顯著升高,而MHC-I則呈升高趨勢(圖1C)。
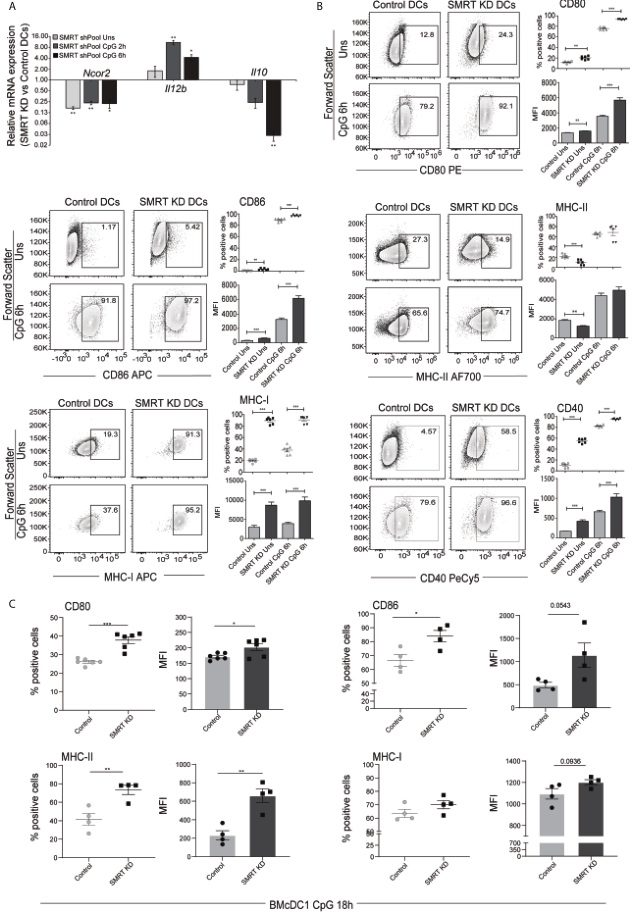
圖1 TLR9與CpG-B配對后SMRT缺失的cDC1和BMcDC1的激活和成熟增強
2、SMRT KD cDC1增加促炎因子IL6,IL-12,IL-23而減少IL-10
接下來,檢測了cDC1系和原發系BMcDC1中重要的DC反應細胞因子的表達。發現與對照cDC1比較,CpG和pIC處理6h后,SMRT敲除顯著增強了陽性細胞的比例和和MFI轉變即IL6,IL-12p40,IL-23p19(圖2A)。在未受刺激和CpG激活6h的SMRT KD cDC1中IL-23p19均顯著升高。相反,與對照組相比,CpG和pIC激活的SMRT KD cDC1上IL-10顯著降低(圖2A)。為了進一步估計這些細胞因子在CpG激活的cDC1系培養液中的水平,進行了生物復合物檢測,發現IL-6、IL-12p40和IL-12p70的分泌水平在SMRT KD cDC1中明顯增加,同時IL-10也隨之減少(圖2B)。與DC中的趨勢相似,與對照組相比,在SMRT KD BMcDC1中觀察到陽性細胞百分比顯著增加,IL-6和IL-23的MFI呈增加趨勢,而IL-10的表達減少。然而,在SMRT KD BMcDC1中IL-12p40的陽性細胞及其MFI雖然高于對照組,但并不顯著(圖2C)。這些結果表明SMRT KD cDC1具有很強的炎癥表型。
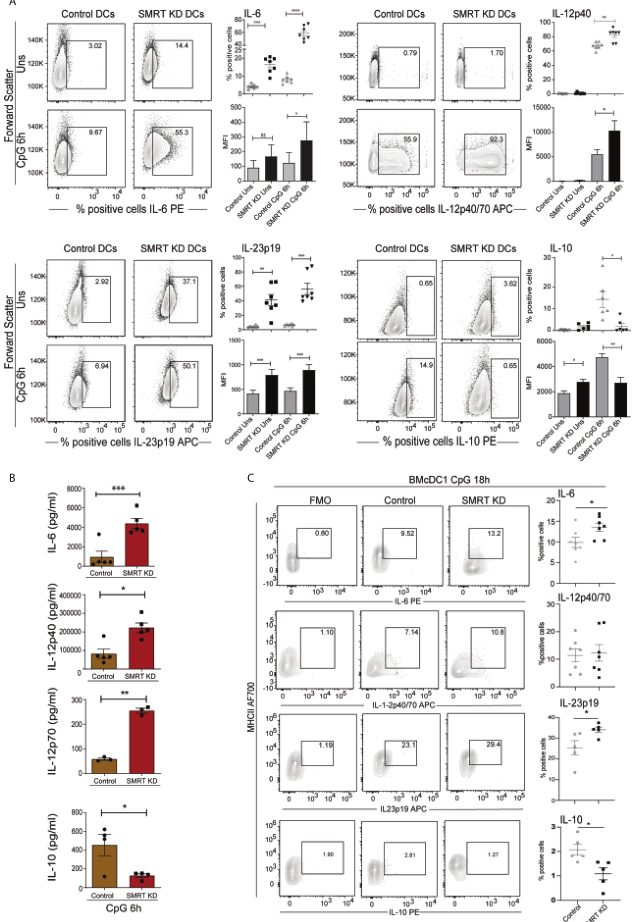
3、OT-II CD4 + Th細胞與SMRT KD cDC1共培養增強Th1和Th17分化
為了解SMRT缺失cDC1對CD4 + Th細胞增殖和分化的功能影響,從OT-II轉基因小鼠脾臟分離純化的CD4 + T細胞與SMRT KD和對照cDC1細胞進行共培養。分離OT-II T細胞,與OVA脈沖和CpG或pIC激活的DC共培養72h檢查增殖,96h檢查分化。在增殖實驗中,我們用efluor-670增殖染料標記OT-II T細胞。
與對照相比,SMRT KD細胞增強T細胞的增殖(圖3A,B)。眾所周知,IL-6抑制FOXP3轉錄因子的表達,從而抑制Treg的分化,并與IL-23一起通過誘導RORgt的表達導致Th17細胞的發育。另一方面,已知IL-12p70上調T-bet導致Th1分化。在OT-II共培養實驗中,發現在CpG激活的SMRT KD cDC1組,CD44+ T-bet + IFN-g +和CD44 + RORgt+ IL-17+細胞的比例增加,其表明Th1和Th17亞型頻率增強的比例顯著高于對照組DC(圖3C-F)。為進一步證實Th17極化增加,檢測了共培養細胞的上清中IL-17細胞因子的分泌水平,發現其水平顯著升高(圖3G)。所有的下游分析都在這些雙陽性細胞中進行。
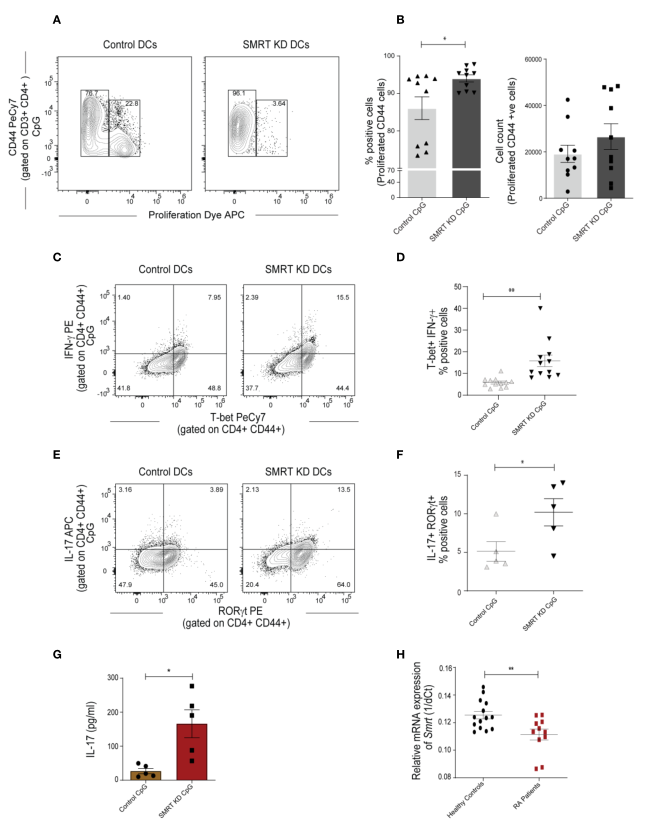
圖3 SMRT KD cDC1增強了Th1和Th17細胞的體外極化
4、類風濕性關節炎(RA)患者的PBMCs的Ncor2表達降低
RA等自身免疫性疾病被廣泛歸類為Th1和Th17疾病,IL-10的表達在這些患者中也被發現大幅降低。早前已經證實Th1反應與自身免疫性疾病有關。然而,對IFN-g和IL-12-/-小鼠的研究表明,這些小鼠有很高的概率發展為膠原誘導關節炎。本研究中,已經確定了Th17細胞在自身免疫中的作用。因此,為確定Ncor2的表達是否在自身免疫性疾病中發生改變,對11例RA患者和14例健康供體的PBMCs進行了Ncor2的RT- qPCR,結果發現與健康對照比較,RA患者PBMCs中Ncor2表達顯著降低(圖3H)。這一結果提示,在RA疾病中Ncor2的降低可能與炎癥表型的增加有關。
5、SMRT KD cDC1與OT-I CD8+ T細胞共培養增加T細胞的細胞毒性
由于觀察到在SMRT耗盡的cDC1中MHC-I表達顯著增加,于是進一步確定這些DC是否有可能增加CD8+ T細胞的細胞毒性。為驗證這一點,從OT-I轉基因小鼠的脾臟中分離出CD8+ T細胞,并將其與對照組和SMRT KD cDC1共培養。首先用SIINFKL (OVA肽257-264)脈沖DC過夜。脈沖DC被CpG或pIC激活2h后與純化的CD8+ T細胞共培養。然后檢測共培養CD8+ T細胞在72h后的增殖和96h后IFN-g、perforin和顆粒酶B(GrB)的表達。使用eFluor 670標記的CD8+ T細胞進行增殖試驗,發現共培養的OT-I CD8+ T細胞增殖未見明顯變化(圖4A,B)。與對照組相比,CpG激活的SMRT KD cDC1共培養的表達IFN-g、perforin和GrB的CD8+ CD44+ T細胞陽性率顯著上調(圖4C)。
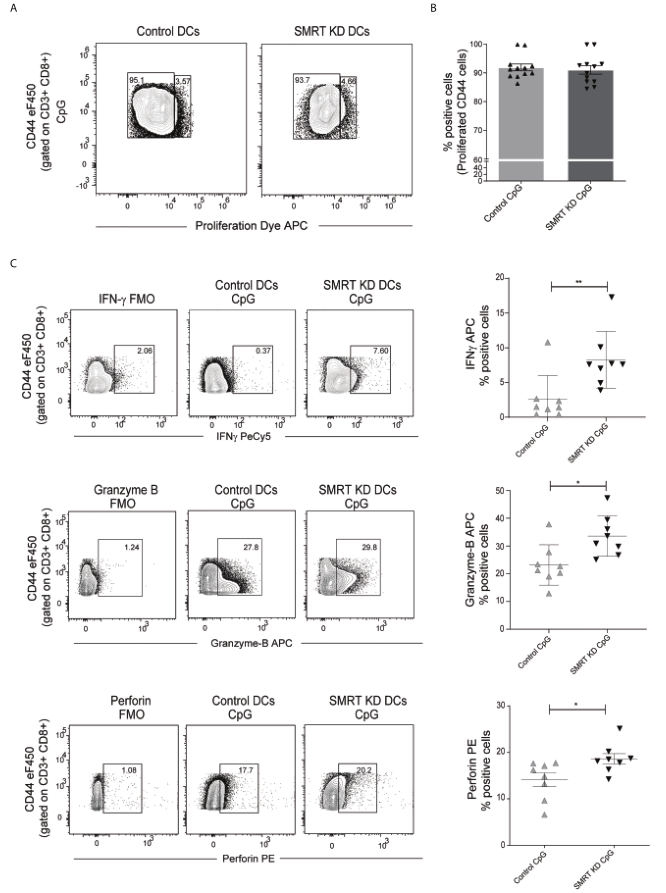
圖4 SMRT KD cDC1增加了CD8 T淋巴細胞體外perforin,granzyme和IFN-g的產生
6、過繼轉移CpG脈沖SMRT缺失的cDC1后,OVA誘導的遲發性超敏(OVA- DTH)小鼠足墊炎癥增強,并且降低小鼠B16F10黑色素瘤負擔
建立OVA- DTH小鼠模型,以研究對照和SMRT KD cDC1 DCs的免疫調節。首先在小鼠耳后皮下用佐劑(明礬alum)乳化的OVA致敏;對照組和SMRT KD cDC1經OVA脈沖4h后經CpG活化2h后于第14天過繼轉移;第20天,DC過繼轉移一周后,小鼠左腳墊局部再次經OVA致敏刺激,以誘導超敏介導的炎癥反應;在OVA再次刺激后每12h至72h測量足墊腫脹,以檢查免疫反應的嚴重程度(圖5A)。結果顯示,與對照組cDC1和PBS處理的動物相比,CpG激活的SMRT KD cDC1處理的小鼠腳墊炎癥明顯增加(圖5B)。接下來,檢查腘窩淋巴結和腹股溝淋巴結中的T細胞亞型。與PBS處理組相比,注射SMRT KD DC的小鼠顯示出明顯增加的Th1亞型,表現為IFN-g和T-bet陽性T細胞數量(圖5C)。同時,注射SMRT KD cDC1的小鼠中Th17 T細胞數量也顯著增加表現為IL-17和RORgt陽性細胞(圖5D)。這些結果表明,DC中SMRT敲低會在宿主體內產生非常強烈的炎癥T細胞反應。
對SMRT KD cDC1增強細胞毒活性的觀察引導進一步研究這些細胞的抗腫瘤潛能。已廣泛報道DC疫苗治療誘導腫瘤相關抗原(TAA)特異性的溶瘤CD8+ T細胞活性。因此,為評估SMRT KD cDC1誘導的細胞毒性CD8 + T細胞增強的生理影響,在C57BL/6小鼠中建立了B16F10黑色素瘤模型。假設,與對照組相比,接種了載有B16抗原的炎性SMRT KD cDC1疫苗的動物能夠抵抗不斷增加的腫瘤負擔。首先,在動物左側皮下接種SMRT KD和對照細胞,這些細胞之前被B16F10細胞裂解液脈沖處理,以誘導對B16F10腫瘤的免疫。3天后,注射強化劑量。強化劑量7天后,在小鼠右側皮下注射106 B16F10細胞(圖5E)。從腫瘤發生后的第7天到第16天,每隔一天測量一次腫瘤體積。與對照組DC和PBS處理組相比,SMRT cDC1組的腫瘤負荷明顯減少(圖5F,G)。為進一步評估CD8+ T細胞的細胞毒性,在腫瘤發生后的第16天殺死小鼠,解剖腫瘤并通過膠原酶處理制成單細胞懸液。用PMA/Ionomycin/BFA刺激這些細胞5小時,用流式細胞儀分析CD8+ T-細胞。與對照組或PBS組相比,SMRT KD cDC1接種動物的效應T細胞的細胞毒性顯著增強,表現為perforin、GzB和IFN-g陽性群體的比率增加(圖5H)。此外,SMRT KD cDC1接種動物腫瘤的重量顯著減輕(圖5I)。
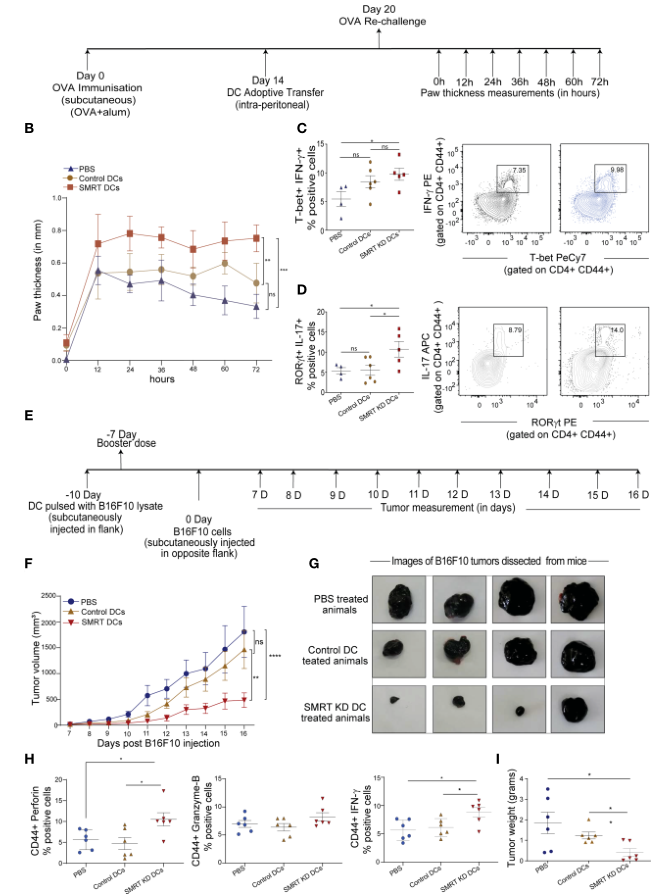
圖5過繼轉移Control和SMRT KD DC的C57BL/6小鼠的DTH反應和腫瘤消退
7、cDC1中NCoR1和SMRT的比較基因組和轉錄組分析顯示,STAT3信號介導的IL- 10調控存在差異
作者此前發現NCoR1直接結合并強烈抑制DC的耐受性基因,如Il10、Il27、Cd83和Socs3。NCoR1敲除急劇增加了這些基因的表達,從而導致耐受性基因的表達,進而導致了Treg的產生。與此相反,本研究發現,NCoR1同源物SMRT KD MutuDC在體外和體內顯著增加炎癥反應。因此,作者進行了NCoR1和SMRT的基因組比較,以及NCoR1和SMRT KD DCs的轉錄組分析,以了解NCoR1和SMRT的不同基因調節。
在CpG刺激6小時下,與對照組比較,SMRT KD cDC1的差異基因表達分析顯示,分別有1273和934個基因上調和下調(圖6A)。通過SMRT的ChIP-seq分析發現,與下調的基因相比,大量的SMRT結合基因被確定為上調,這支持了其作為輔抑制因子的作用(圖6B)。此外,在SMRT KD條件下,直接上調靶基因的IPA分析顯示炎癥反應途徑的上調,如IL-12信號通路(圖6C)。然而,與NCoR1相反,SMRT KD顯示IL-10信號通路在CpG刺激6h時下調(圖6D)。
為識別NCoR1和SMRT KD后表現出不同表達模式的基因集,對DEGs進行無監督K-means聚類,并確定了6個聚類(圖6E)。cluster-1中的基因在NCoR1和SMRT KD中均表現出成倍增加的變化,并參與豐富的通路,如“Interferon signaling”,“Activation of IRF by cytosolic pattern recognition receptors”,“IL-12 signaling and production in macrophages”。相反,cluster-2基因在SMRT和NCoR1 KD 6h CpG條件下表現出差異調控,富集的通路為“Th2 pathway”,“STAT3 pathway”,“IL-17-A signaling”和“IL-10 Signaling”(圖6F)。這些觀察結果清楚地表明,炎癥免疫反應基因如Il12b和Il6均被NCoR1和SMRT抑制,而調控基因如Il10、Socs3僅被NCoR1強烈抑制(圖6G)。
此外,NCoR1和SMRT峰的差異結合鑒定出NCoR1主導(12,473)、普通NCoR1-SMRT主導(5949)和SMRT主導(2707)基因組區域(圖6H, I)。與NCoR1顯性區和常見的NCoR1-SMRT結合區主要分布在內含子或遠端基因間區相比,SMRT主導區在啟動子-近端區域占優勢,(圖6H)。這進一步表明,SMRT主要通過啟動子-近端結合調控基因,而NCoR1和常見的NCoR1-SMRT結合基因則通過遠端調控元件調控。
此外,與基因組區域相關的基因KEGG通路分析顯示,NCoR1或常見NCoR1-SMRT結合的CpG依賴性增加主要富集“Th1 and Th2 pathway”,“Th-17 signaling pathway”,“NFkB signaling pathway”,“JAK-STAT signaling pathway”(圖6J)。CpG激活后,NCoR1和SMRT在TSS的幾個近端或遠端區域共同占據炎癥(Il12b)和耐受基因(Il10),表明這兩種抑制因子都參與調節免疫反應基因(圖6K)。
為確定在這些輔抑制子結合的基因組區域在CpG激活6小時前后募集的轉錄因子,進行de novo基序富集分析,發現PU.1、RUNX2和Jun-Fos/AP1轉錄因子基序在幾乎所有NCoR1都富集,而SMRT結合的基因組區域在未受刺激或CpG激活6小時時富集。有趣的是,NFkB基序在CpG顯性NCoR1、顯性SMRT和普通NCoR1-SMRT結合位點被富集。在未受刺激和6h CpG激活條件下,IRF8和IRF4基序分別在顯性NCoR1和常見NCoR1- SMRT區域富集(圖6L)。
除上述基序外,還發現在CpG顯性NCoR1-SMRT和CpG顯性SMRT基因組區STAT3基序富集。由于NFkB家族成員和Stat3轉錄因子在調節炎癥(Il12b, Il6)和耐受(Il10, Socs3)基因表達中起著重要作用,為進一步了解耐受基因的下調,檢測了與Jak-Stat信號通路相關的其他基因的表達。發現其他幾種Jak-Stat信號的調控存在明顯差異,SMRT KD DC中Jak-Stat信號被下調而NCoR1 KD DC中無變化(圖7A, B)。總的來說,基因組和轉錄組比較分析表明,與NCoR1 KD DC相比,SMRT KD DC顯示STAT3-IL-10軸失調。
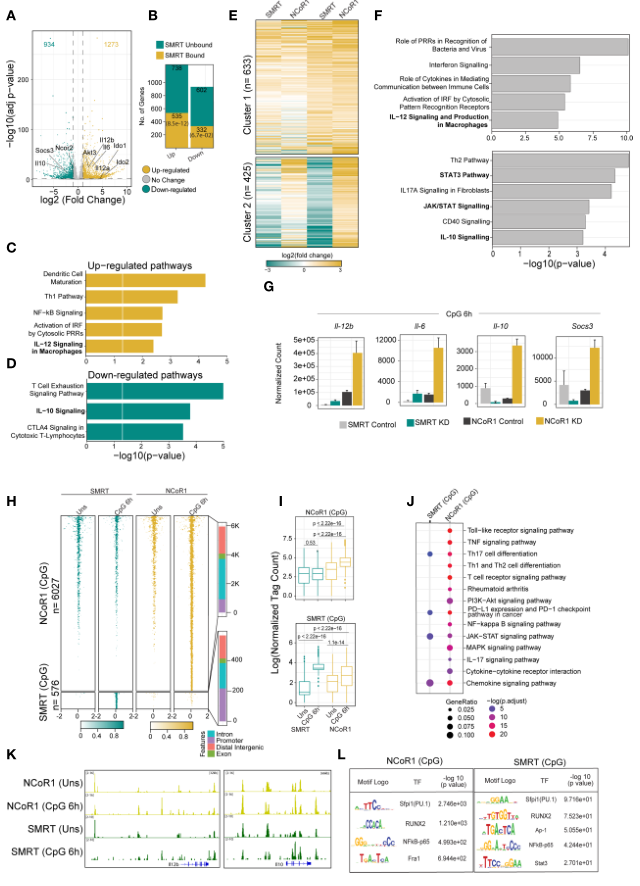
圖6整合基因組學分析(RNA-seq和ChIP-seq)鑒定SMRT和NCoR1在cDC1免疫應答調節中的差異作用
8、SMRT介導的NR4A1下調抑制mTOR-STAT3信號,導致IL-10抑制
STAT3 TF在Jak-Stat信號通路和調控Il10和Socs3的表達中起著核心作用。在SMRT KD cDC1中,NFkB抑制基因Nfkbia和Tnfaip3在CpG刺激6h后也被下調。首先,為確認NCoR1和SMRT中p-STAT3的差異調控,在CpG激活后0h、2h和6h對NCoR1 KD、SMRT KD和對照cDC1中的p-STAT3進行WB。結果顯示,與對照細胞相比,SMRT KD cDC1中p-STAT3表達下調,而在NCoR1 KD DC中p-STAT3表達上調(圖7C, D)。
據報道,STAT3結合Il10基因調控其表達。所以作者對p-STAT3進行染色質免疫沉淀(ChIP),然后進行RT-qPCR,以推斷對照組、NCoR1 KD和SMRT KD cDC1在CpG處理2h后p-STAT3與Il10基因的結合情況。觀察到,相對于對照DC,SMRT KD DC中p-STAT3與Il10的結合減少,而另一方面,與對照DC相比,NCoR1 KD細胞中p-STAT3與Il10的結合增強(圖7E)。
此外,為了解這些DC中STAT3信號的上游控制,作者查閱了相關文獻,發現mTOR可控制STAT3激活,另一方面mTOR受核受體NR4A1(又叫Nur77)調控。首先檢測NCoR1和SMRT敲低cDC1中p-mTOR的調控,發現在NCoR1 KD cDC1中p-mTOR上調,而在SMRT敲低后p-mTOR急劇降低(圖7F,G)。此外,在SMRT和NCoR1缺失DC中Nr4a1 (Nur77)的差異調控。SMRT KD導致Nr4a1表達下調,而NCoR1 KD對Nr4a1表達無顯著影響(圖7H)。使用IGV檢測了ChIP-seq數據中NCoR1和SMRT在Nr4a1上的直接結合,觀察到在CpG激活6h后,SMRT而不是NCoR1結合TSS轉錄本(圖7H)。作者還通過評估在CpG激活2h和6h前后SMRT KD和對照cDC1中的NR4A1蛋白表達來證實這一結果:與對照細胞相比,SMRT KD DC中的NR4A1蛋白表達顯著下調(圖7I, J)。
為進一步證實NR4A1確實在SMRT KD細胞中通過mTOR控制STAT3信號,使用NR4A1慢病毒構建了穩定的NR4A1 KD和空載體cDC1細胞,以確認其在mTOR- STAT3 - IL-10信號中的作用。在NR4A1敲除效率通過WB證實(圖7K, L)。在CpG激活2h后檢測p-mTOR和p-STAT3,發現與對照細胞相比,NR4A1缺失的cDC1中p-mTOR和p-STAT3顯著減少(圖7K, L)。此外,在NR4A1缺失的cDC1中,CpG處理6h后IL-10陽性細胞顯著減少,相應的MFI也減少(圖7M)。此外,作者還嘗試在SMRT KD cDC1中短暫過表達NR4A1 ORF,但由于過表達質粒轉導后細胞狀態不佳,未能成功進行這一分析,因此,使用了另一種方法。報道稱6- MP可誘導細胞NR4A1的表達。因此,使用6-MP增強SMRT KD cDC1中Nr4a1的表達,看看它是否可以補充IL-10的表達。結果顯示6-MP處理增強SMRT KD cDC1中Nr4a1的表達,導致IL-10的表達增加(圖7N)。這些結果證實,SMRT KD介導的NR4A1下調導致STAT3激活減少,從而降低IL-10水平,增強cDC1的炎癥表型。
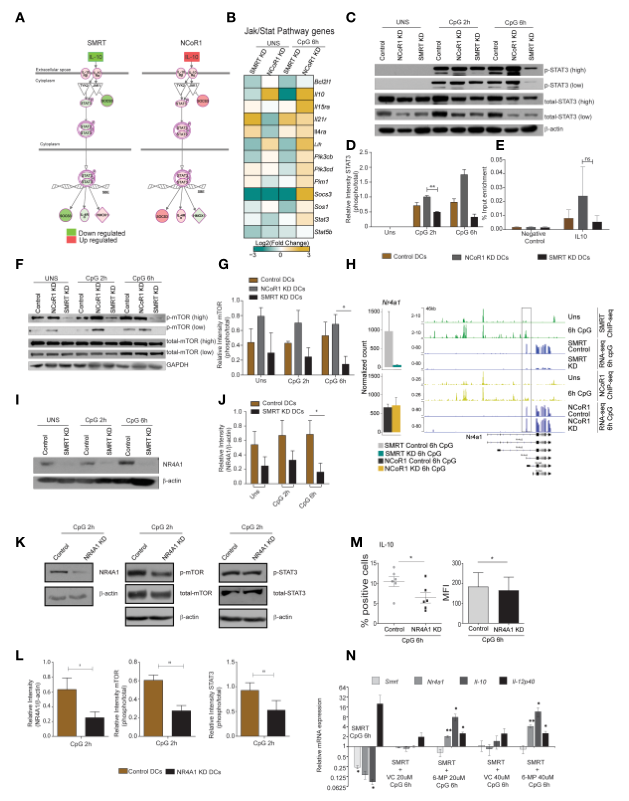
圖7 Nurr-77、mTOR、Stat3信號調節IL-10在SMRT - KD cDCs中的表達
參考文獻:
Jha A, Ahad A, Mishra GP, Sen K, Smita S, Minz AP, Biswas VK, Tripathy A, Senapati SB, Gupta B, Acha-Orbea H and Raghav SK (2022) SMRT and NCoR1 fine-tune inflammatory versus tolerogenic balance in dendritic cells by differentially regulating STAT3 signaling. Front. Immunol. 13:910705. doi: 10.3389/fimmu.2022.910705