






一個新的長鏈非編碼RNA SP100-AS1通過海綿化miR-622和穩定ATG3誘導結直腸癌放射抵抗
雖然放療是治療結直腸癌(CRC)的重要手段,但臨床上放療抵抗的發生率仍然很高。長鏈非編碼RNA (lncRNA)通過在轉錄或翻譯后水平調控基因或蛋白在結直腸癌放療抵抗中發揮重要作用。該研究旨在鑒定與放射抗拒相關的新型lncRNA。該研究發表于《Cell Death & Differentiation》,IF:12.067。
技術路線:
主要研究結果:
1. SP100-AS1表達上調與結直腸癌放療抵抗相關
采用RNA-seq分析比較8例放療敏感和8例放療抵抗患者結直腸癌組織中lncRNA的差異表達(圖1A)。結果表明,與輻射敏感樣本相比,267個lncRNA在輻射抗拒樣本中表達上調(圖1B),其中SP100-AS1顯著過表達。接下來,RT-qPCR分析表明,SP100-AS1在CRC組織(n = 44)中的表達顯著高于癌旁正常組織(圖1C)。綜上所述,上述結果表明SP100-AS1可能在CRC進展中發揮重要作用。此外,研究者假設SP100-AS1可能通過其在放射抗拒組織中的異常表達調控CRC的放射敏感性。因此,研究者收集了具有放射敏感和放射抗拒臨床特征的結直腸癌患者的組織。如圖1D所示,放射抗拒的結直腸癌組織中SP100-AS1的表達量較高。綜上所述,研究者的研究結果表明,SP100-AS1參與CRC的進展和放射抵抗。采用RT-qPCR檢測正常人結腸黏膜上皮細胞NCM460和結直腸癌細胞株LS177T、HCT116、CT26、HCT8、HT29、LoVo和SW480中內源性SP100-AS1的表達(圖1E)。結果表明,與正常上皮細胞株相比,CRC細胞中SP100-AS1的表達水平較高,尤其是在HCT116和SW480細胞株中,并用于后續實驗。有趣的是,SP100-AS1的高表達與結直腸癌患者的不良生存相關(圖1F)。綜上所述,這些發現表明lncRNA SP100-AS1的過表達與CRC的放射抵抗和低生存率相關。
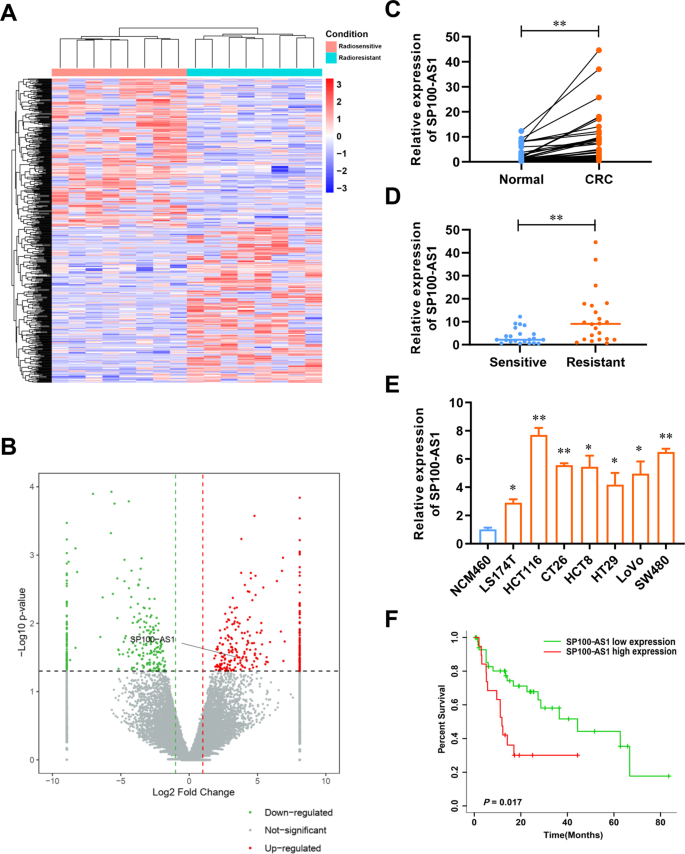
圖1 SP100-AS1在CRC中表達上調,且與放療呈正相關
2. 敲低SP100-AS1可增強結直腸癌的放射敏感性
在建立了SP100-AS1在CRC患者組織和細胞系中的異常表達模式后,研究者研究了這種lncRNA如何調節腫瘤細胞的增殖和生存。如前所述,SP100-AS1在HCT116和SW480細胞中以劑量依賴性方式表達最多(圖1E)。克隆形成實驗檢測SP100-AS1對HCT116和SW480細胞放射敏感性的影響。值得注意的是,當SP100-AS1表達下調時,這兩種細胞的放射敏感性顯著增加(圖2A, C)。此外,研究者對HCT116和SW480細胞在4 Gy劑量下的活力進行了研究,發現沉默SP100-AS1降低了細胞的增殖和活力(圖2B, D)。
放射治療引起的DNA雙鏈斷裂是公認的,必須在腫瘤進一步生長之前進行修復。這一發現強調需要開發一種新的方法來阻止DNA修復和阻止腫瘤生長,或至少減緩它們以延長患者的生存期。阻斷DNA損傷修復是治療CRC的一種特別有吸引力的策略,因為CRC對輻射具有高度抵抗力。正如預期的那樣,抑制SP100-AS1可以誘導γ-H2AX的表達,γ-H2AX是DNA損傷的顯著標志物(圖2E)。接下來,作者分析SP100-AS1對細胞凋亡的影響。Annexin V/PI雙染色顯示,與對照組相比,SP100-AS1缺失顯著增加了HCT116細胞的凋亡反應(圖2F)。這表明SP100-AS1參與了CRC輻射誘導的DNA損傷和細胞凋亡。因此,敲低SP100-AS1可以增加CRC的放射敏感性。
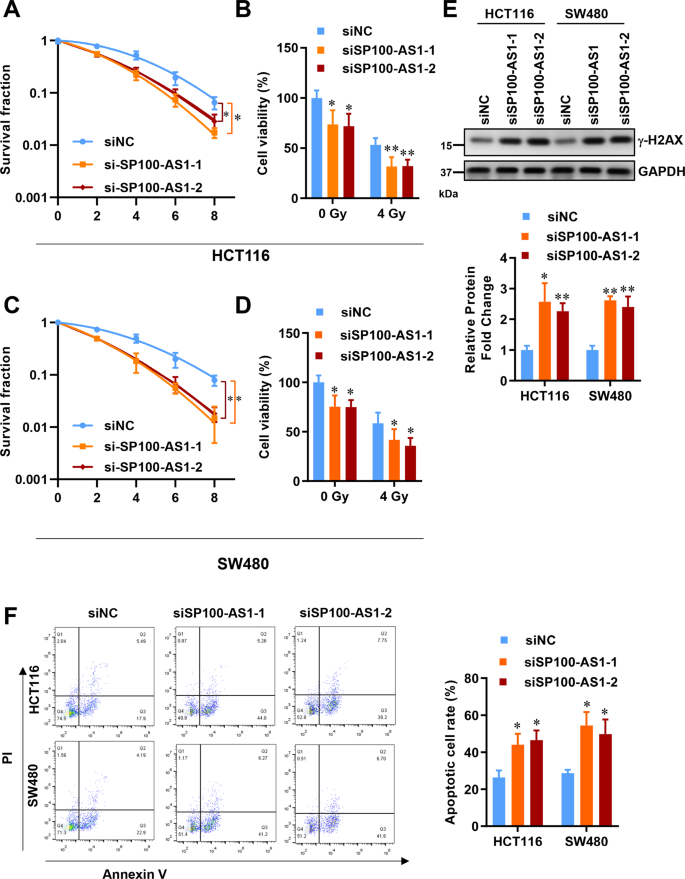
圖2 SP100-AS1下調加重了輻射誘導的細胞死亡
通過建立BALB/c裸鼠移植瘤模型,研究SP100-AS1在體內的作用。如圖3A所示,通過慢病毒感染HCT116細胞,SP100-AS1被穩定敲低,接種的異種移植瘤比對照組的腫瘤生長減慢。此外,IR后,與對照組相比,SP100 - AS1敲低的HCT116腫瘤細胞生長緩慢(圖3A)。分割IR后,沉默SP100-AS1可進一步減緩CRC腫瘤的生長(圖3B)。實驗結束后,測量腫瘤重量(圖3C)。隨后,轉移酶介導的dUTP缺口末端標記(TUNEL)實驗表明,敲低SP100-AS1在2 Gy輻射下引起更多的DNA損傷,并降低Ki67染色,表明SP100-AS1下調抑制CRC的生長速度,尤其是在暴露于輻射時(圖3D)。綜上所述,這些結果表明,SP100-AS1沉默可以在體內外增強CRC的放射敏感性。
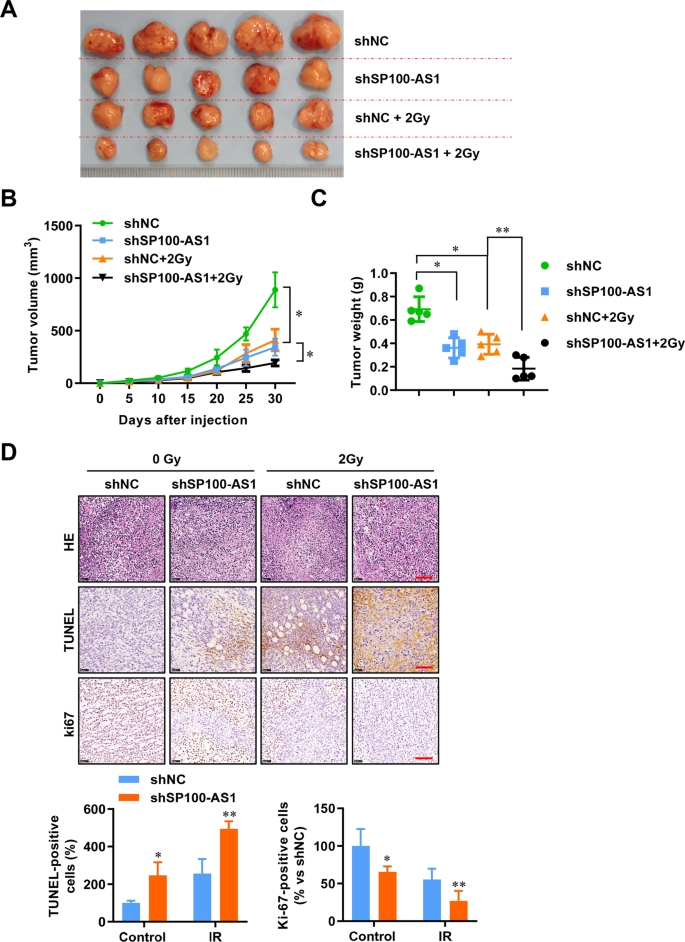
圖3 SP100-AS1下調降低了受照結直腸癌細胞的體內生長
4. SP100-AS1通過自噬途徑增加結直腸癌細胞的放射抵抗活性
自噬通路的激活在包括結直腸癌在內的多種腫瘤的放射抵抗中發揮重要作用。為了探究自噬通路是否參與HCT116和SW480細胞的輻射抗性,作者定量分析了敲低SP100-AS1后,自噬流標記物LC3-II和p62/SQSTM1的蛋白表達。結果顯示LC3-II降低,p62蛋白水平升高(圖4A)。此外,作者使用LC3-GFP-RFP報告系統來評估自噬體形成。LC3是一種與綠色和紅色熒光蛋白融合的膜結合蛋白。與RFP不同,GFP在酸性環境中不穩定。相應計算RFP點位或RFP/GFP比值來測量自噬通量。敲低SP100-AS1后,用過表達LC3-GFP-RFP的慢病毒感染HCT116細胞,并給予相應水平的輻射處理。與之前的研究結果一致,與對照組相比,放射治療對自噬的誘導作用要弱得多,自噬流顯著減少(圖4B)。此外,western blot和IHC分析表明,在IR處理的CRC異種移植瘤中,p62蛋白表達顯著降低,LC3-II蛋白水平顯著升高,而沉默SP100-AS1可以逆轉這一情況。因此,這些結果得出結論,輻射可以誘導HCT116細胞的自噬,沉默SP100-AS1可以緩解這一效應。

圖4 SP100-AS1通過自噬途徑調控HCT116細胞增殖
5. SP100-AS1通過ATG5和Beclin1調控自噬通路
ATG5和Beclin1是宏觀自噬通路的重要上游調控因子,過表達ATG5和Beclin1可直接激活自噬信號通路。因此,作者研究了SP100 - AS1調控的自噬是否可以通過過表達ATG5和Beclin1來拯救。在HCT116和SW480細胞中敲低SP100-AS1后,ATG5和Beclin1表達上調,并檢測LC3-II的蛋白表達。如圖5A所示,SP100-AS1下調導致的自噬流減少被ATG5或Beclin1上調所恢復。此外,Annexin V/PI雙染后采用流式細胞術定量凋亡,表明上調ATG5和Beclin1可減少敲低SP100-AS1誘導的細胞凋亡(圖5B)。如前所述,使用LC3-GFP-RFP慢病毒報告檢測自噬流密度,并測量每個細胞的紅色點狀點數量。在HCT116細胞中,上調ATG5或Beclin1可以顯著增加SP100-AS1敲低后的點狀細胞數量(圖5C)。此外,作者研究了ATG5和Beclin1對HCT116和SW480細胞增殖和存活的功能和作用。這些結果表明,在這兩個細胞系中,ATG5或Beclin1上調均可挽救SP100-AS1下調導致的細胞增殖下降(圖5D),這意味著SP100-AS1通過經典的宏觀自噬途徑調節自噬。

圖5 ATG5和Beclin1過表達挽救了SP100-AS1對自噬的調節作用
6. SP100-AS1與ATG3相互作用并調節其蛋白穩定性
lncRNA可以與特定的蛋白質相互作用來發揮其功能。通過熒光原位雜交(FISH)檢測SP100-AS1在HCT116和SW480細胞中的亞細胞定位,探討SP100-AS1是否能通過這一機制發揮作用。令人驚訝的是,SP100-AS1在細胞質中表現出強烈的熒光,這表明該lncRNA可以與細胞質中的蛋白結合并進行相關功能,包括細胞增殖和輻射抗性(圖6A, B, C)。接下來,作者進行了RNA下拉實驗,以發現與SP100-AS1相互作用的蛋白。如圖6D所示,銀染法在分子量35 kDa處檢測到多條條帶(圖6D)。質譜分析顯示ATG3肽水平顯著升高。進一步對SP100-AS1進行RIP實驗,western blot結果顯示SP100-AS1能與ATG3特異性結合(圖6E)。另一方面,使用抗atg3抗體鑒定相關的結合RNA,結果顯示,與對照IgG組相比,SP100-AS1在atg3結合的提取液中顯著富集(圖6F)。嚴格地說,對SP100-AS1下拉試驗中提取的蛋白進行的蛋白質印跡分析證實,ATG3蛋白與SP100-AS1的義序列特異性結合,但與反義對照無特異性結合(圖6G)。綜上所述,作者發現ATG3是SP100-AS1在體內和體外的特異性結合蛋白。
在確定了SP100-AS1可以與ATG3相互作用之后,作者接下來試圖研究SP100-AS1是否可以調節ATG3蛋白的穩定性或功能。首先,作者在HCT116細胞中檢測敲低SP100-AS1后ATG3的蛋白水平。下調和過表達SP100-AS1分別可顯著降低和升高ATG3蛋白水平(圖6H)。蛋白質穩定性受多種途徑和機制調控,尤其是泛素化依賴的蛋白酶體降解信號通路。與此一致的是,ATG3可以泛素化。因此,作者評估了SP100-AS1是否可以通過蛋白酶體途徑影響ATG3蛋白的穩定性。在HCT116細胞中,轉染Flag-tagged ATG3后,HA標記的泛素蛋白過表達,而內源性SP100-AS1 lncRNA被敲低。抗flag抗體免疫沉淀后,用抗HA抗體檢測ATG3泛素化水平。作者發現,沉默SP100-AS1增加了ATG3的泛素化水平,表明SP100-AS1介導的ATG3降解參與了泛素化依賴性降解(圖6I)。此外,作者還研究了ATG3在放線菌酮(cycloheximide, CHX)(一種成熟的蛋白質合成抑制劑)處理下的降解過程。重要的是,作者發現敲低SP100-AS1可以加速ATG3的降解(圖6J)。在HCT116細胞中敲低或過表達SP100-AS1,然后用蛋白酶體抑制劑MG132處理。如圖6K所示,敲低SP100-AS1引起的ATG3蛋白降解被挽救。此外,過表達SP100-AS1和MG132處理后,ATG3的表達顯著增加。這些結果證實了SP100-AS1與ATG3相互作用并調節其蛋白穩定性。
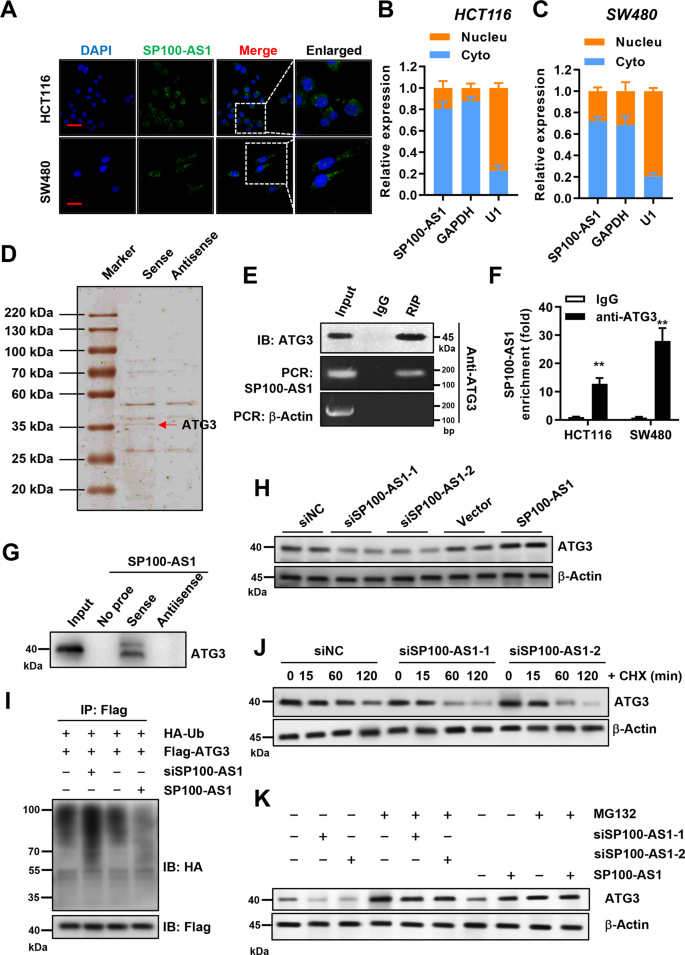
圖6 SP100-AS1與ATG3相互作用并調節其蛋白穩定性。
7. SP100-AS1在結直腸癌中可作為miR-622的海綿
為了進一步研究SP100-AS1調控ATG3蛋白表達的機制,作者評估了SP100-AS1是否可以作為ATG3 mRNA的miRNA海綿。通過RIP實驗確定AGO2是否與SP100-AS1相關,AGO2在miRNA通路中發揮多種作用,并通過產生pre-miRNA參與miRNA組裝過程。有趣的是,在HCT116和SW480細胞系中均檢測到AGO2和SP100-AS1之間存在顯著的相互作用(圖7A, B)。接下來,作者試圖確定哪些miRNA可以直接與SP100-AS1結合。作者使用lncRNA探針篩選幾個miRNA與SP100-AS1之間的相互作用。采用靶向SP100-AS1的特異性探針,通過pull-down實驗純化與SP100-AS1相關的miRNA。結果顯示,SP100-AS1和miR-622的富集最為顯著(圖7C)。這些發現證實了miR-622和SP100-AS1在HCT116細胞中的相互作用。如圖7D所示,SP100-AS1可以與miR-622顯著結合。綜上所述,這些發現表明SP100-AS1在CRC中扮演了miR-622海綿的角色。
生物信息學分析發現SP100-AS1的3'UTR區含有miR-622的靶向基序。構建熒光素酶實驗系統檢測miR-622對SP100-AS1 mRNA的調控作用(圖7D)。螢火蟲熒光素酶盒連接到野生型或突變的SP100-AS1 3'UTR區域。作者發現,突變的SP100-AS1 3'UTR不能與miR-622相互作用(圖7E)。熒光素酶質粒轉染HCT116細胞。過表達miR-622時,攜帶野生型區域的熒光素酶強度低于突變型(圖7F)。相反,抑制miR-622 (miR-622-in)誘導熒光素酶活性顯著增加,這一活性也被結合位點突變所抑制(圖7G)。綜上所述,作者證實了miR-622可以直接結合SP100-AS1并影響其在CRC細胞中的mRNA穩定性。

圖7 SP100-AS1在結直腸癌中充當miR-622的海綿
8. miR-622靶向ATG3 mRNA調控自噬流
考慮到SP100-AS1海綿化miR-622并下調自噬流,作者試圖研究miR-622是否影響自噬通路。在HCT116細胞中過表達miR-622或SP100-AS1。有趣的是,在SP100-AS1過表達的HCT116細胞中,異位表達miR-622可以恢復p62和LC3-11的表達(圖8A, B)。因此,作者假設SP100-AS1可以作為miR-622的海綿,阻止其對ATG3 mRNA的影響,從而穩定ATG3,促進自噬流。采用計算機模擬方法分析ATG3的3'UTR區域以驗證這一假設。出人意料的是,作者檢測到了miR-622的一個靶向基序。接下來,作者進行了熒光素酶測定,將螢火蟲熒光素酶盒插入野生型和突變的ATG3 3'UTR區域的前面(圖8C)。如圖8D所示,過表達miR-622可以降低野生型熒光素酶的表達,而共轉染SP100-AS1可以挽救熒光素酶的活性。相反,共轉染攜帶突變ATG3 3'UTR區域的質粒不能恢復熒光素酶的活性(圖8D)。然后研究HCT116細胞中ATG3的內源性表達,結果顯示miR-622在mRNA和蛋白水平顯著降低ATG3的表達,而與SP100-AS1共表達可以恢復ATG3的表達。正如預期的那樣,使用siRNA敲低SP100-AS1也可以降低ATG3 mRNA水平(圖8E、F、G)。

圖8 SP100-AS1通過與miR-622相互作用調控ATG3表達
結論:
該研究表明SP100-AS1/miR-622/ATG3軸有助于CRC患者的放射抵抗和自噬活性,提示其作為改善CRC放射治療反應的治療靶點具有巨大的前景。
參考文獻:
Zhou Y, Shao Y, Hu W, Zhang J, Shi Y, Kong X, Jiang J. A novel long noncoding RNA SP100-AS1 induces radioresistance of colorectal cancer via sponging miR-622 and stabilizing ATG3. Cell Death Differ. 2022 Aug 17. doi: 10.1038/s41418-022-01049-1. Epub ahead of print. PMID: 35978049.