






VEGF糖基化參與血管生成
N-糖基化和二硫鍵的形成是蛋白質折疊的兩個重要步驟,它們發生在內質網(ER)中,相互影響。本研究為分析N-糖基化和氧化之間的相互作用,研究了蛋白質二硫氧化酶ERO1-α如何影響血管生成VEGF121的糖基化,VEGF121是血管穩態的關鍵調節因子。本研究揭示ERO1丟失對蛋白質N-糖基化的影響不僅與血管生成有關,而且與其他癌癥病理機制有關。本研究于2022年10月發表在《Redox Biology》IF:10.787期刊上。
技術路線:
實驗結果:
1、ERO1敲除的細胞氧化能力減弱氧化鍵形成受損
為了跟蹤WT和ERO1 KO HeLa細胞中ER的氧化還原平衡,利用比例氧化還原傳感器ER定位的roGFP2。如圖1A所示,在WT和ERO1 KO細胞中,DTT刺激后ER roGFP2迅速減少;然而,在還原劑的洗脫過程中,ERO1 KO細胞表現出較慢的恢復速度,而WT細胞的氧化平衡恢復得很快,ERO1 KO細胞在40分鐘內未達到WT細胞的氧化平衡基線值,這表明缺乏ERO1會損害ER的氧化平衡。
為評估ERO1缺失對ER中二硫鍵形成速率的影響,作者構建了FLAG-VEGF121,它被組裝成二硫連接的同型二聚體(圖1B),并比較了在WT和ERO1 KO細胞中,在蛋白合成抑制劑CHX脈沖作用后,細胞內形成二硫連接的同型二聚體的速率。結果顯示,FLAG-VEGF121在WT和ERO1 KO細胞中的積累程度相似;然而,雖然在WT細胞中二聚體是所有時間點的主要物質,但在ERO1 KO細胞中,二聚體的生成速度較慢,單體在沖洗過程中積累,表明單體轉化為二聚體的速度較慢(圖 1C)。 因此,由ERO1損失引起的氧化平衡的改變轉化為 VEGF121 二硫鍵形成速率的損害。

圖1ERO1 KO細胞中氧化能力受損和VEGF121二硫鍵形成延遲
2、ERO1 KO細胞分泌的VEGF121無鏈內二硫鍵的缺陷而是N75上高糖基化
為分析WT和ERO1 KO HeLa細胞分泌的FLAG-VEGF121,收集兩細胞系的條件培養基,用N -乙基馬來酰亞胺(NEM)烷基化,然后用FLAG-M1抗體免疫沉淀。免疫沉淀采用非還原SDS-PAGE分析。經考馬斯藍染色發現WT細胞的FLAG-VEGF121有三個主要條帶,對應于VEGF121的二聚體和寡聚體,而ERO1 KO細胞的FLAG-VEGF121只有兩個條帶,即一個微弱的寡聚體和一個比WT細胞分泌的慢的遷移二聚體(圖2A)。從凝膠中分離這些條帶,DTT還原并用碘乙酰胺(IAA)烷基化,這一處理導致二硫鍵中半胱氨酸被標記,導致氨基甲基(CAM)富集,最后進行nLC-ESI-MS/MS(圖2B)。裂解肽的質譜顯示,WT和ERO1 KO分泌的FLAG-VEGF121在NEM和IAA肽烷基化方面沒有太大差異。例如,參與VEGF121鏈內二硫鍵的C68只以氧化形式存在于WT和ERO1 KO細胞的VEGF121中;C102也參與VEGF121的鏈內二硫鍵,在WT和ERO1 KO細胞的VEGF121中均以混合還原和氧化狀態檢測到,這表明在ERO1 KO細胞中FLAG-VEGF121的分子內二硫鍵沒有缺陷(圖2B)。
使用NetNGlyc在線軟件預測VEGF121 氨基酸序列的共有序列 NIT 內75號位處存在N-糖基化位點。因此,作者假設圖2A遷移較慢的條帶是N-糖基化的產物,并使用糖苷內切酶對此進行測試。如圖2C所示,WT和ERO1 KO細胞的FLAG-VEGF121對內糖苷酶H (ENDO H)不敏感,但對肽- N -糖苷酶F (PNGase F)敏感,導致快速遷移的蛋白質消失。這些結果表明,遷移較慢的多肽含有N -鏈寡糖,在高爾基體中已經成熟,從而獲得了對EndoH的抗性。用PNGase F(一種酰胺酶)處理后,糖基化Asn殘基被脫酰胺。對WT和ERO1 KO細胞中FLAG-VEGF121二聚體和寡聚體的片段肽進行質譜分析,然后用PNGase F酶切,證實天冬酰胺N75被糖基化,除了來自WT細胞的低聚物2(圖2D)。挽救實驗證實了ERO1在限制糖基化中的作用。在 ERO1 KO 細胞中轉染ERO1或其高活性突變體ERO1C131A的表達質粒,通過改變糖基化形式占總量的 80% 至 40%,拯救了ERO1 KO細胞分泌的FLAG-VEGF121的N-高糖基化(圖2E)。

圖2 ERO1 KO細胞分泌的VEGF121鏈內二硫鍵和N-高糖基化
3、ERO1 KO細胞VEGF121的N75高糖基化導致分泌動力學缺陷
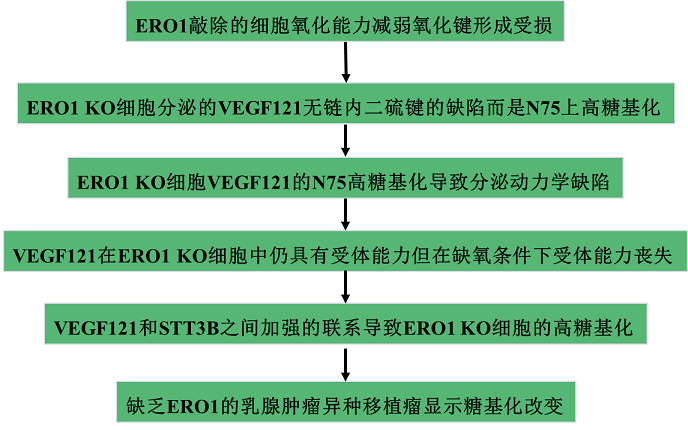
接下來比較VEGF121在WT和ERO1 KO細胞的分泌動力學,并研究N-高糖基化如何影響這一過程。結果發現ERO1 KO細胞分泌VEGF121的動力學受損,在最后兩個時間點達到平臺期(圖3A)。為研究該損傷是否由于高糖基化,構建一個突變體,其中N75被Gln殘基(N75Q)取代。發現VEGF121突變體在WT和ERO1 KO細胞中表達時都沒有糖基化(圖3B)。值得注意的是,該突變體在WT和ERO1 KO細胞中的分泌動力學相當,而在ERO1 KO細胞中的分泌受影響較小(圖3C-3D),表明N75的高糖基化是ERO1 KO細胞中FLAG-VEGF121分泌受損的基礎。
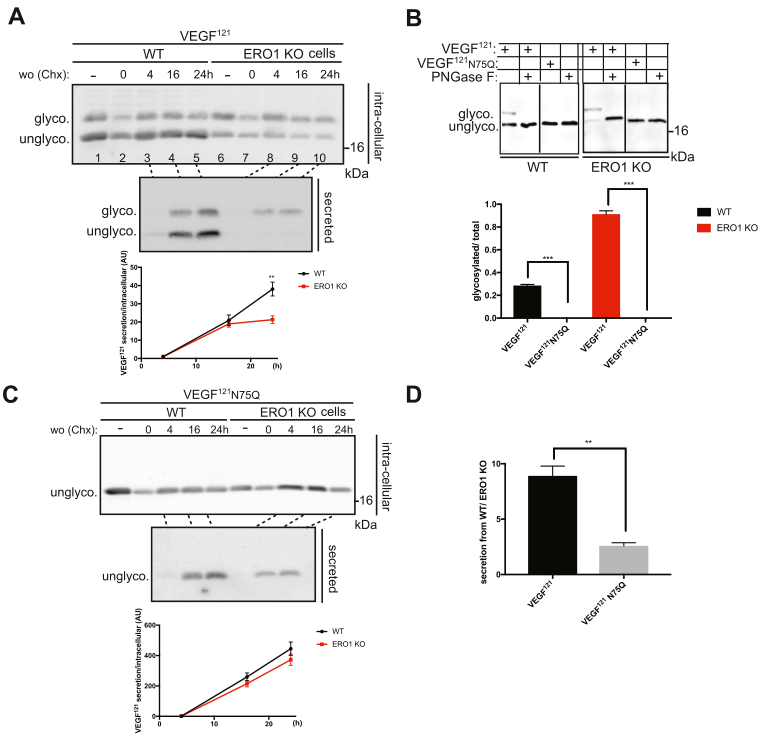
圖3 ERO1 KO細胞分泌的VEGF121的N75高糖基化損害其其分泌動力學
4、VEGF121在ERO1 KO細胞中仍具有受體能力但在缺氧條件下受體能力喪失
突變為FLAG-VEGF121的C60 (C60S)或C68 (C68S)絲氨酸,分別參與鏈間和鏈內二硫鍵(圖1B),增加了WT細胞分泌的VEGF121的N-糖基化,表明VEGF中二硫鍵形成的受損改善了其N -糖基化(圖4A)。與未突變的VEGF121相比,WT細胞中C60的分泌不受影響,而C68的分泌受損60%(圖4A)。當ERO1 KO細胞分泌兩種突變體C60和C68時,未觀察到高糖基化,表明ERO1缺失是VEGF121糖基化的主要影響。與相同細胞中未突變的VEGF121相比,ERO1 KO細胞中C60的分泌不受影響,而C68的分泌受損85%,WT細胞中C68的分泌受損更嚴重(圖4A)。
利用表面等離子體共振(SPR)來確定Aflibercept的不同形式VEGF121的相對解離常數(KD),這是一種親和力的測量方法,Aflibercept是人IgG1的Fc區與VEGF兩受體VEGFR1和R2之間的融合蛋白。結果表明,ERO1 KO細胞分泌的VEGF121對受體的親和力大約是WT細胞分泌的VEGF121的2倍(但無統計學差異),但突變的VEGF121 C60S的親和力是WT細胞分泌的VEGF121 C68S的6倍,而突變的VEGF121 C68S則完全不與受體結合。表明,來自ERO1 KO細胞的VEGF121仍然具有受體活性,排除了其大規模氧化展開的可能(圖4B)。
在低氧條件下,WT細胞分泌的VEGF121在糖基化或分泌量方面與正常缺氧條件下分泌的VEGF121相比,差異可以忽略不計(圖4C)。此外,其KD在數量上與常氧條件下分泌的對應物相似(圖4D)。相反,在缺氧條件下,ERO1 KO細胞中VEGF121的分泌量受到嚴重影響(圖4C),這表明在ERO1缺失的條件下,VEGF121的成熟和分泌特別依賴氧氣。此外,KD降低兩倍表明缺氧條件下ERO1 KO細胞VEGF121受體能力進一步受損(圖4D)。
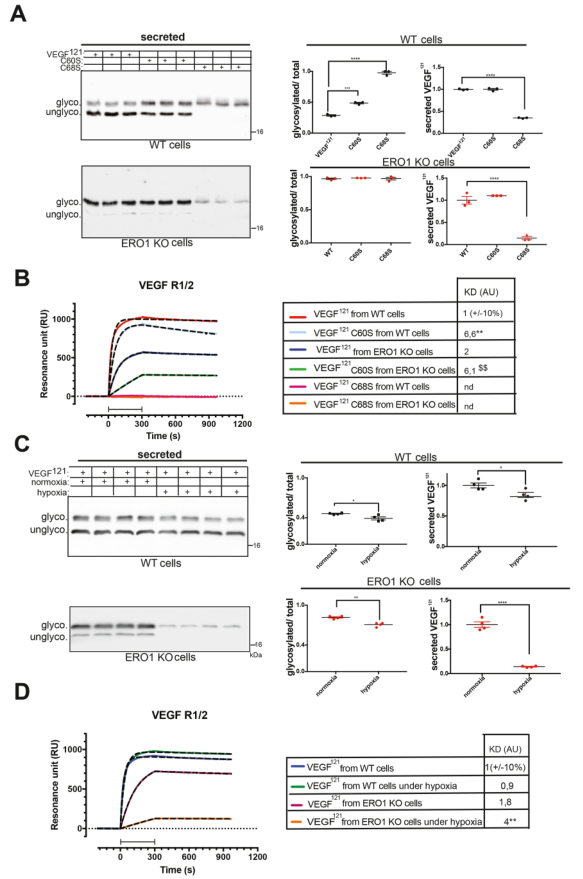
圖4 ERO1 KO細胞的VEGF121具有受體能力但在缺氧條件下其受體能力受損
5、VEGF121和STT3B之間加強的聯系導致ERO1 KO細胞的高糖基化
正如已經觀察到的,在ERO1 KO細胞中,細胞內VEGF121以及分泌形式都是高糖基化的。然而,與分泌型VEGF121不同的是,N-糖基化的細胞內形式對EndoH和PNGaseF都敏感(圖5A)。在WT和ERO1 KO細胞中,VEGF121對兩種內糖苷酶的敏感性沒有差異,這表明在穩定狀態下,兩種細胞系中大多數細胞內VEGF121都沒有通過高爾基體過渡。細胞內FLAG-VEGF121 C68S和C68S的表現與WT細胞中高糖基化的分泌對應物非常相似,與ERO1 KO中的原生形式沒有相關差異(圖5B)。為從分子層面深入了解VEGF121高糖基化的原因和/或后果,分析了VEGF121在WT和ERO1 KO細胞中的相互作用物。轉染VEGF121、VEGF121C60S和VEGF121C68S的WT和ERO1 KO細胞的蛋白裂解物用FLAG M1抗體免疫沉淀后SDS-PAGE電泳。在考馬斯藍凝膠中,可以看到貫穿泳道的一系列條帶(圖5C)。

圖5 在ERO1 KO和半胱氨酸突變體中細胞內VEGF121高糖基化
對圖5C的每個泳道進行單獨的切除和消化,將洗脫后的多肽進行nLC-ESI MS/MS分析,從而鑒定出VEGF121相互作用分子。對ERO1 KO細胞中VEGF121、VEGF121C60S和VEGF121C68S相互作用分子的通路分析表明,主要富集至通過天冬酰胺進行N-鏈糖基化的通路(圖6A)。研究發現,在ERO1 KO細胞中,含有硫氧還蛋白結構域的STT3B復合物亞基MAGT1與所有三個VEGF121相互作用(圖6B)。基于此,作者決定關注VEGF121C68S,因為C68中的突變是最接近糖基化Asn的,因此,其糖基化應該受MAGT1影響較小。免疫沉淀證實,VEGF121和VEGF121C68S在WT和ERO1 KO細胞中均存在MAGT1免疫反應帶(圖6C)。用MAGT1-HA轉染的WT和ERO1 KO細胞的NEM烷基化允許在非還原免疫印跡上解析 MAGT1 的不同氧化還原形式。來自ERO1 KO細胞的MAGT1出現在大約52 kDa的緩慢遷移帶中,這在 WT 細胞中不可見,表明 MAGT1 在 ERO1 KO 細胞中的氧化還原介導的相互作用(圖6D)。在WT和ERO1 KO細胞中,MAGT1和STT3B的缺失導致VEGF121和VEGF121C68S的低糖基化,表明VEGF121及其突變體都是STT3B/MAGT1的底物(圖6E)。然而,VEGF121C68S(最接近天冬酰胺的半胱氨酸突變體)的N-糖基化對 STT3B/MAGT1干擾不敏感。這些發現表明,ERO1缺失通過MAGT1增強了STT3B與VEGF121底物之間的相互作用,導致其N-高糖基化。
圖6 ERO1-KO細胞中VEGF121及其半胱氨酸突變體與MAGT1的相互作用增強
6、缺乏ERO1的乳腺腫瘤異種移植瘤顯示糖基化改變
為探究ERO1在腫瘤中的生理性影響,構建ERO1缺失或未缺失的三陰性乳腺腫瘤模型。用兩種具有不同糖特異性的凝集素:小麥胚芽凝集素(WGA)和隔離素B4(IB4)對異種移植物切片進行染色。結果如圖7所示,雖然IB4染色在兩種基因分化腫瘤中表現相似,但ERO1 KO細胞中的WGA染色平均比WT細胞高1.5倍,并在KO細胞中表現出斑片狀聚集模式。對這些腫瘤簇所占體積的定量分析顯示KO腫瘤與WT腫瘤相比增加了5倍,表明ERO1缺陷在調節糖基化方面具有廣泛的作用。
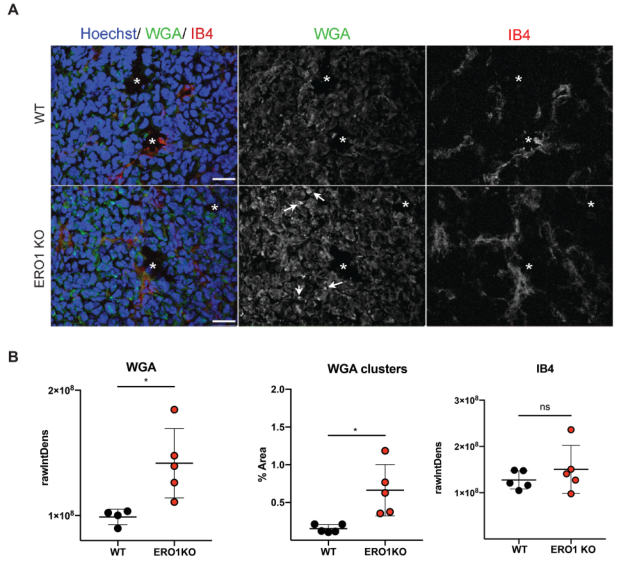
圖7 ERO1-KO乳腺腫瘤表現為N-高-糖基化蛋白模式
綜上所述,本研究證實ERO1缺失會導致較少的氧化ER氧化還原平衡。這導致 VEGF121中二硫鍵形成延遲,這有利于其N-高糖基化并增加MAGT1捕獲潛力,進一步增加其在N75上的糖基化。這反過來減慢了ERO1耗盡細胞中生長因子的分泌,如圖8所示。
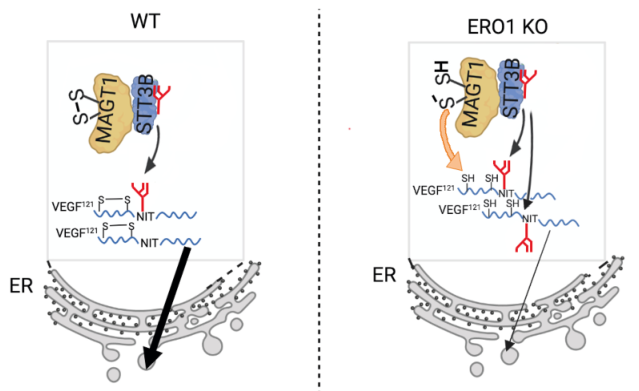
圖5 機制模式圖
參考文獻:
Varone Ersilia., Chernorudskiy Alexander., Cherubini Alessandro., Cattaneo Angela., Bachi Angela., Fumagalli Stefano., Erol Gizem., Gobbi Marco., Lenardo Michael J., Borgese Nica., Zito Ester.(2022). ERO1 alpha deficiency impairs angiogenesis by increasing N-glycosylation of a proangiogenic VEGFA. Redox Biol, 56(undefined), 102455. doi:10.1016/j.redox.2022.102455