






線粒體聯動鐵死亡保衛心臟的新策略
細胞鐵死亡是由鐵依賴性脂質過氧化作用驅動的調節性細胞死亡的一種形式。線粒體在細胞鐵死亡中發揮著重要的作用,也與一系列疾病顯著相關。線粒體GSH(mitoGSH)庫的過度耗竭或氧化關聯到多種病理生理條件。本研究發現FUNDC2通過調控mitoGSH來調節鐵死亡應激。研究強調通過阻斷鐵死亡和改善mitoGSH庫和功能來保護DOX誘導的心肌病。這可能成為一種保護心臟的新策略。本研究于2022年9月發表在《PNAS》IF:12.779期刊上。
技術路線:

主要研究結果:
作者首先通過WB分析篩選主要器官中FUNDC2的表達情況,發現FUNDC2在心臟中高表達,暗示它的潛在功能(圖1A)。也發現,當個體完全丟失FUNDC2蛋白后(圖1B),其后代的表型符合孟德爾定理而且正常生長。另外,FUNDC2敲除(FUNDC2-KO)的成年小鼠擁有正常的體重和健康的心臟功能(圖1C-G)。接著作者給野生型小鼠和FUNDC2-KO小鼠分別注射一劑10 mg/kg DOX(生理鹽水作為對照),結果發現與野生型小鼠相比,FUNDC2-KO小鼠的體重和心臟重量顯著顯著減少(圖1C)。經胸壁超聲心動圖通過測量左心室射血分數(LVEF)和左心室縮短率(LVFS)用來評估心臟功能(圖1D)。注射生理鹽水出的WT型小鼠和FUNDC2-KO小鼠在LVEP和LVFS上均沒有明顯的變化,但是與空白對照WT型小鼠相比,用DOX處理過FUNDC2-KO小鼠的心臟功能被保護地更好(圖1E)。用DOX處理WT小鼠的心房利鈉肽(Anp)、腦鈉肽(Bnp)、肌球蛋白7(Myh7)等生物標志物的mRNA水平都顯著上調,而鹽處理組沒有差異(圖1F)。這些心力衰竭生物標志物的mRNA水平也支持這一觀點。Masson ' s Trichrome染色表明FUNDC2的敲除可以防止DOX誘導的心臟纖維化(圖1G)。這些結果表明喪失FUNDC2保護小鼠免受DOX誘導的心肌病。
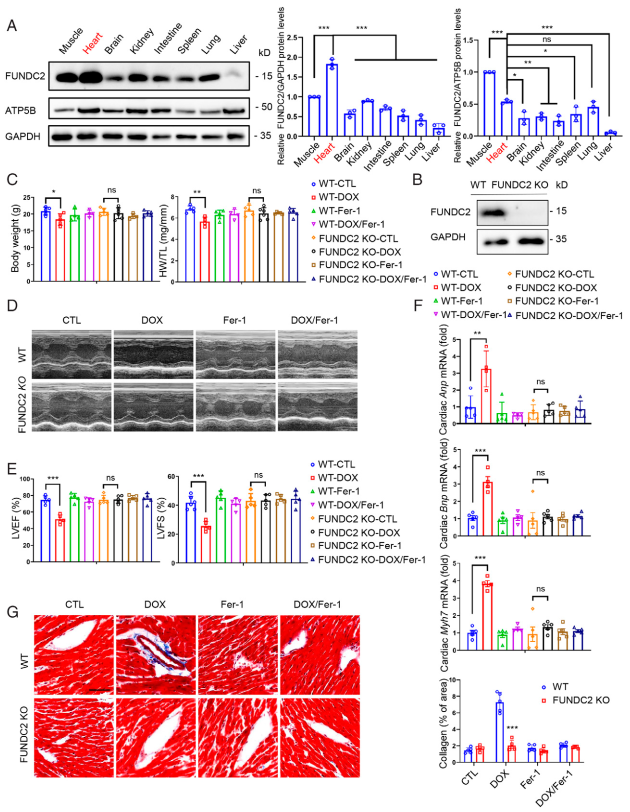
圖1敲除FUNDC2可改善DOX誘導的心肌病
作者推測FUNDC2通過調節細胞鐵死亡影響DOX誘導的心臟中毒。作者首先驗證Fer - 1預處理對WT和FUNDC2-KO兩種小鼠心臟損傷都有很強的保護作用(圖1C-G)。接著進一步探索FUNDC2在DOX誘導的鐵死亡中的作用。結果發現,DOX誘導WT小鼠心臟的Ptgs2 mRNA表達顯著上調近3倍,而Fer - 1預處理后上調作用被抑制(圖2A)。然而,在KO小鼠中沒有觀察到Ptgs2 mRNA表達的變化(圖2A)。
作者進一步檢測脂質過氧化的生物標志物,4 - 羥基壬烯醛(4-HNE)的水平,這也是體內鐵死亡的關鍵指標。結果發現4 - HNE水平在DOX處理的WT小鼠心臟組織中顯著增加(圖2B-C),但在FUNDC2 - KO小鼠中沒有。作者還分析脂質過氧化產物丙二醛(MDA)的水平,發現其上調只能在WT小鼠中檢測到,而在FUNDC2 - KO小鼠中檢測不到(圖2D,左)。作者隨后分離心臟組織的亞細胞組分,發現DOX處理增強線粒體中的MDA水平,但不在細胞質中(圖2D,中間,右)。值得注意的是,MDA只在野生型小鼠中增加,不在FUNDC2 - KO小鼠中增加(圖2D,中間,右)。作者的結果與DOX處理特異性增強線粒體脂質過氧化的觀察一致。相反,FUNDC2缺失可以阻止DOX誘導的脂質過氧化增加。作者進一步使用電子顯微鏡檢查線粒體形態,發現DOX處理心臟中的線粒體破裂,嵴扭曲,嵴密度降低(圖2E)。相比之下,FUNDC2的缺失在很大程度上阻止這種線粒體損傷。最后,Fer - 1強烈抑制DOX誘導的線粒體變形,暗示DOX -線粒體-鐵死亡信號軸。

圖2敲除FUNDC2緩和DOX誘導的鐵死亡。
3、FUNDC2 - KO抑制Erastin誘導的細胞死亡
GSH代謝處于氧化還原代謝和鐵死亡的中心。作者檢測心臟組織中總GSH / 谷胱甘肽二硫化物(GSSG)比值和GSH水平,發現FUNDC2 - KO心肌中GSH / GSSG比值和GSH水平遠高于WT組(圖3A)。由于FUNDC2是一種線粒體蛋白,作者試圖確定FUNDC2是否調節線粒體GSH。事實上,FUNDC2 - KO心臟中線粒體GSH / GSSG比值和GSH水平高于WT組(圖3A)。值得注意的是,在WT心臟中,總的和線粒體的GSH / GSSG比值和GSH水平都被DOX處理降低到50 %,但在KO心臟中保存(圖3A)。這些數據表明,在DOX誘導的心臟中毒中,高GSH水平,特別是mitoGSH,可能具有心臟保護作用。
為進一步理解FUNDC2管理mitoGSH的分子機制,作者用WT和FUNDC2-KO MEF細胞進行體外研究。與體內結果一致,FUNDC2 - KO MEF細胞中線粒體和胞漿GSH / GSSG比值和GSH (cytoGSH)水平顯著高于WT MEF細胞(圖3B)。Erastin處理降低WT和FUNDC2 - KO MEF細胞的GSH / GSSG比值和GSH水平,但FUNDC2 - KO MEF細胞的GSH / GSSG比值和GSH水平在所有條件下均高于WT MEF細胞(圖3B)。GSH水平與erastin誘導的細胞死亡相關(圖3C),表明GSH在鐵死亡中的功能重要性。使用脂質過氧化的熒光探針C11 - BODIPY通過流式細胞術檢測脂質ROS的積累,證明FUNDC2的缺失阻止erastin誘導的脂質ROS積累(圖3D)。DOX誘導的鐵死亡也在MEF細胞中得到證實。
為進一步證實FUNDC2通過mitoGSH促進鐵死亡,作者用mitoGSH處理MEF細胞,并用一種特定的mitoGSH消耗化學物質mitoCDNB選擇性損耗mitoGSH。mitoCDNB預處理降低線粒體GSH / GSSG比值和GSH水平,erastin處理后進一步降低(圖3E)。mitoCDNB處理有效增強erastin誘導的WT細胞和FUNDC2- KO MEF細胞死亡和脂質ROS積累(圖3F-H)。這些結果表明mitoGSH耗竭確實增加erastin誘導的細胞死亡。

圖3FUNDC2-KO抑制但mitoGSH耗竭增加erastin誘導的細胞死亡。
作者研究FUNDC2與SLC25A11之間的相互作用是否在鐵死亡中發揮功能。,作者首先以scramble shRNA作為對照,在WT和FUNDC2 - KO MEF細胞中穩定敲低SLC25A11(圖4A),發現SLC25A11缺失降低WT和FUNDC2 - KO MEF細胞中線粒體GSH / GSSG比率和GSH水平,而對胞漿GSH / GSSG比率和GSH水平無明顯影響(圖4B)。SLC25A11 KD也增強erastin誘導的鐵死亡,無論FUNDC2水平如何,細胞活力,細胞死亡(圖4C-E)和脂質ROS積累都證明了這一點(圖4F)。這些數據表明SLC25A11對于細胞抵抗鐵死亡是必需的。
作者進一步探索在SLC25A11中獲得功能是否可以防止erastin誘導的鐵死亡。將編碼載體或Flag - SLC25A11的質粒穩定轉染到WT和FUNDC2 - KO MEF細胞中(圖4G),發現在WT和FUNDC2 - KO MEF細胞中,無論是否用erastin處理,SLC25A11過表達均增加線粒體GSH / GSSG比值和GSH水平(圖4H)。因此,過表達SLC25A11足以抑制erastin誘導的細胞死亡(圖4I-K)和脂質ROS積累(圖4L)。這些結果表明SLC25A11可以對抗FUNDC2依賴性的鐵死亡。

圖4 FUNDC2–SLC25A11軸調節鐵死亡
在這里作者探究FUNDC2如何調控SLC25A11。通過WB檢測,作者發現FUNDC2-KO可以增加SLC25A11水平,并阻斷erastin誘導的GPX4和線粒體蛋白如TIM23和TOM20在MEF細胞中的減少(圖5A)。此外,FUNDC2 - KO小鼠心臟組織中SLC25A11水平高于WT小鼠(圖5B)。沒有觀察到SLC25A10的影響,另一個SLC25家族成員(圖5A-B)。盡管在WT和FUNDC2 - KO小鼠中,線粒體蛋白和GPX4在響應DOX時均下調,但FUNDC2 - KO緩解下調的程度(圖5B)。然后作者測試在鐵死亡過程中FUNDC2和SLC25A11之間的相互作用是否增強。通過免疫共沉淀實驗,結果發現erastin處理增強FUNDC2和SLC25A11之間的相互作用(圖5C)。類似的結果在體內心臟組織中得到重現(圖5D)。
因為許多線粒體蛋白需要組裝成寡聚體結構才發揮其功能,作者通過凝膠電泳檢測SLC25A11的寡聚體狀態。如圖5E所示,在MEF細胞中,用溫和的非變性洗滌劑Nonident P - 40裂解線粒體時,SLC25A11蛋白以二聚體形式存在;而在強變性洗滌劑十二烷基硫酸鈉(SDS)裂解時,只能觀察到SLC25A11的單體形式。無論erastin處理與否,FUNDC2-KO MEF細胞中SLC25A11的二聚體和單體蛋白水平均高于WT MEF細胞(圖5E)。同樣,在心臟組織和它們在體內的線粒體部分中也可以觀察到差異,盡管程度相對較小(圖5F)。這些結果表明FUNDC2通過調節SLC25A11的穩定性和二聚化來調節其功能。
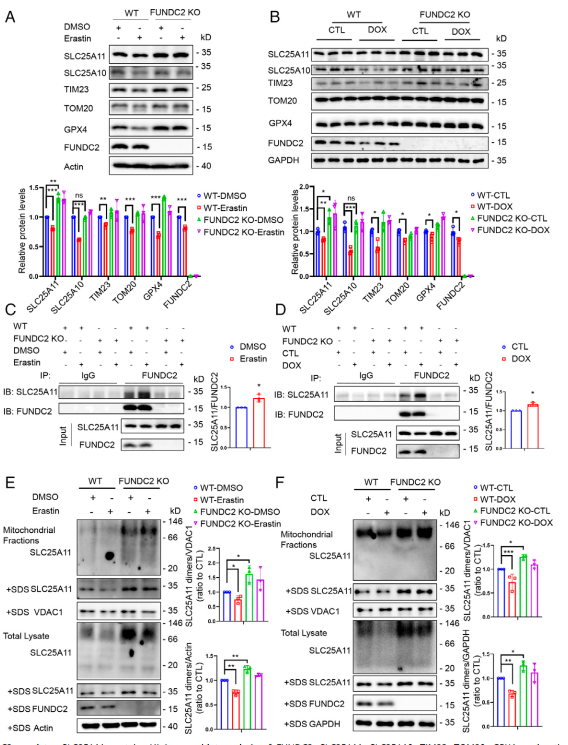
圖5 FUNDC2調控SLC25A11蛋白
結論
本研究結果表明線粒體FUNDC2對于DOX誘導的心肌病是重要的。敲除FUNDC2在很大程度上預防DOX誘導的心肌病。進一步研究發現,FUNDC2調控mitoGSH以及DOX誘導的鐵死亡和心臟損傷需要穩定的GPX4和SLC25A11。也說明線粒體定位的GPX4抑制脂質過氧化的產生,并防止DOX誘導的鐵死亡。本研究強調通過阻斷鐵死亡和改善mitoGSH庫和功能來保護DOX誘導的心肌病的策略。
參考文獻
Ta N, Qu C, Wu H, Zhang D, Sun T, Li Y, Wang J, Wang X, Tang T, Chen Q, Liu L. (2022). Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA. 119(36):e2117396119. doi: 10.1073/pnas.2117396119.