






線粒體 交聯 鐵死亡 在心肌病中的作用
線粒體氧化磷酸化(OXPHOS)系統缺陷引起的線粒體疾病的常見表型是心肌病和心衰。線粒體蛋白酶通過降解錯誤折疊的蛋白質和平衡線粒體動力學和線粒體自噬來控制線粒體的適應性。線粒體內膜(IM)蛋白酶Oma1在線粒體應激時被激活,如線粒體去極化、氧化應激、錯誤折疊的IM蛋白質積累、或熱應激。本研究發現OMA1-DELE1介導的綜合應急反應(ISR)在線粒體心肌病中具有保護作用,并將鐵死亡與OXPHOS缺乏和線粒體疾病聯系起來。本研究于2022年9月發表在《Cell Metabolism》IF:31.373期刊上。
技術路線如下:

主要結果如下:
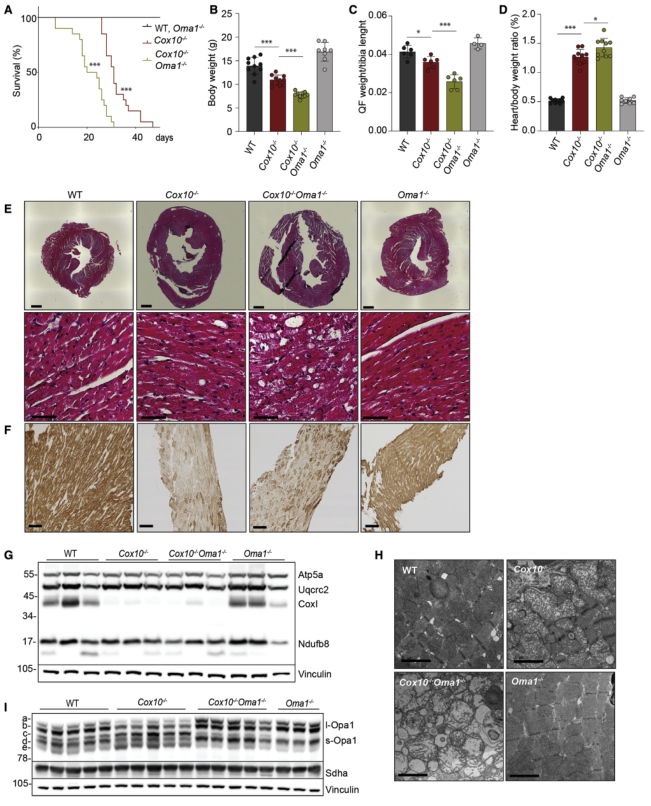
為檢測Oma1在線粒體OXPHOS疾病中的作用,在肌酸激酶啟動子(Ckmm-Cre)的控制下,用表達Cre重組酶的小鼠繁殖Cox10fl/fl和Oma1fl/fl,從骨骼肌和心肌中敲除Cox10和Oma1基因。如圖1A,心臟和骨骼肌中Cox10缺失導致小鼠早發擴張型心肌病和死亡(中位年齡31天)。Cox10?/?小鼠表現為生長遲緩,骨骼肌量進行性減少,心臟腫大,并伴有輕度結締組織積累(圖1B-E)。如預期的,觀察到COX亞基和組裝的COX復合體的水平急劇下降,心臟COX活性降低(圖1F-G) 。通過透射電子顯微鏡觀察到心肌組織的線粒體碎裂,線粒體超微結構改變,線粒體腫大,嵴破裂(圖1H)。心臟中Cox10的缺失導致了Oma1的激活,表現為Opa1處理的增加和Opa1短形式c和e的積累(圖1I)。
此外,Oma1的缺失使Cox10-/-小鼠的心臟表型惡化,而單獨的Oma1-/-缺失沒有明顯影響。Cox10-/-Oma1-/-小鼠的壽命較短,中位生存期為 22 天(圖 1A),并且表現出更嚴重的心肌病和纖維化病變,表明細胞死亡(圖 1B-1E)。Cox10-/-小鼠中Oma1的缺失穩定了長Opa1形式并減少了線粒體碎片(圖1I)。然而,在缺乏Oma1的Cox10-/-心臟中,COX 亞基和組裝的COX復合物的積累沒有受到影響,嵴形態也沒有恢復(圖 1F-1H)。這些實驗揭示Oma1在體內OXPHOS 缺乏中的保護作用。
OXPHOS缺乏可通過多種機制損害各種小鼠組織和細胞模型的溶酶體功能。與此報道一致,在Cox10?/?心臟中觀察到含有線粒體和膜聚集物的大型溶酶體結構(圖2A-C),并觀察到溶酶體標記物Lamp1和自噬底物p62/SQSTM1(簡稱p62)的積累(圖2D-E)。與Cox10?/?心臟相比,缺陷溶酶體結構在Cox10-/-Oma1-/-心臟中進一步積累,并且在Oma1缺失后Lamp1和p62的穩態水平顯著增加(圖2F-G)。這些結果表明,在Cox10?/?缺陷的心肌細胞中,線粒體功能障礙導致溶酶體缺陷,且在缺少Oma1的情況下進一步惡化。
由于觀察到pS6和p4E-BP1、mTORC1底物在Cox10?/?和Cox10-/-Oma1-/-心臟中積累(圖-2H-I),所以進一步檢查了用雷帕霉素抑制mTORC1后是否會延長小鼠的壽命。結果顯示雷帕霉素抑制Cox10?/?和Cox10-/-Oma1-/-心臟S6磷酸化(圖2H-I)。然而,用雷帕霉素喂養不影響小鼠的存活(圖2J)、線粒體超微結構(圖 2K)、p62和Lamp1的積累(圖2H-I)。 因此,得出結論,增加mTORC1信號傳導不會導致這些小鼠的溶酶體缺陷,并且Oma1通過另一種機制延長Cox10?/?小鼠的壽命。
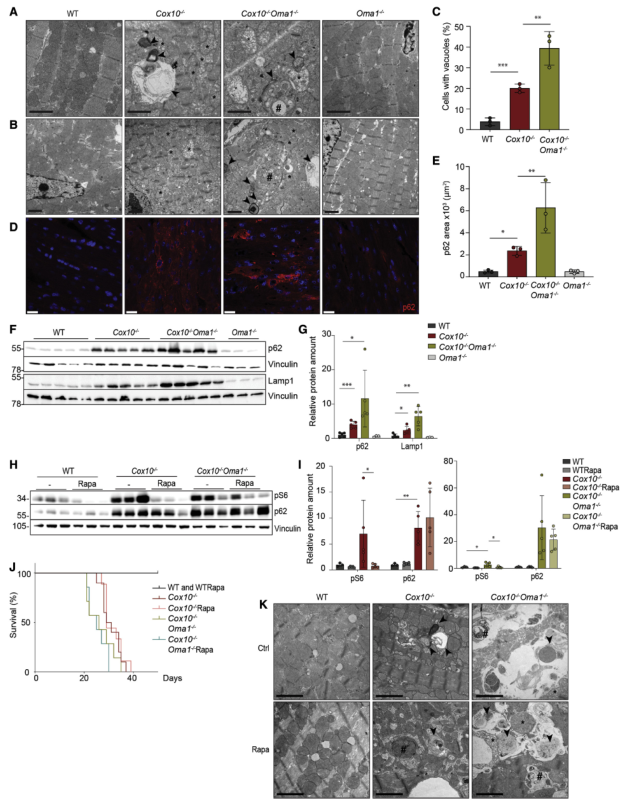
圖2 線粒體心肌病中Oma1缺失導致溶酶體缺陷和p62積累
3、Oma1激活Cox10?/?小鼠心臟的ISR
對4周大野生型、Cox10?/?、Oma1?/?、Cox10?/? Oma1?/?小鼠心臟組織進行測序并對差異表達基因進行KEGG分析。結果鑒定到Nrf2信號通路是Cox10?/?心臟中最顯著誘導的通路(圖3A)。根據靶基因表達,p53、Atf4和Nrf2是在Cox10?/?心臟中被預測為最顯著改變的轉錄因子(圖3B)。比較Cox10?/?和Cox10?/? Oma1?/?小鼠心臟的轉錄組,發現Atf4是變化最顯著的轉錄因子(圖3C)。根據文獻報道,作為ISR的一部分,Atf4和其它轉錄因子一起介導eIF2α信號通路可響應不同的線粒體損傷。與Cox10?/? Oma1?/?心臟的ISR受損一致,在Cox10?/? Oma1?/?小鼠心臟中,Atf4靶基因Fgf21、Pycr1和Mthfd2的表達明顯低于Cox10?/?小鼠(圖3D)。對上述4種小鼠的TMT無偏蛋白組分析證實了以上發現(圖3E)。免疫印跡證實,與Cox10?/?心臟相比,Cox10?/? Oma1?/?心臟中Atf4和eIF2α磷酸化水平降低(圖3F-G)。綜上,在心臟OXPHOS缺乏的反應中,Oma1是誘導Atf4介導ISR所必需的。
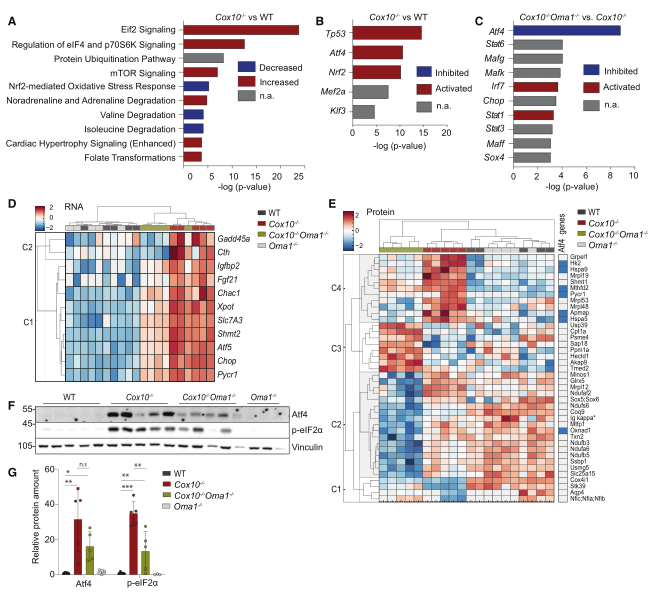
圖3 Cox10?/?小鼠心肌表現出Nrf2和p53激活以及Oma1和Dele1介導的Atf4信號通路
4、Cox10?/?小鼠中ISR誘導是Dele1依賴的
作者在此前的實驗中為探究Oma1作用ISR激活的底物,比較了4種小鼠的線粒體蛋白組,分析發現Oma1促進了Cox10?/?心臟中可能未組裝的COX亞基的蛋白水解,,其蛋白水解可能介導ISR激活,但沒有發現候選Oma1底物。雖然在心臟中表達,但作者通過質譜沒有檢測到Dele1,Dele1已被確定為Oma1底物和Atf4依賴性線粒體應激信號的關鍵成分。因此,構建Dele1?/?小鼠,并將其與心臟和肌肉特異性Cox10?/?小鼠雜交,以研究Dele1在體內對ISR信號的作用。敲除Dele1對小鼠的生存沒有廣泛影響,直到1歲時,在Dele1缺失的情況下,小鼠都沒有表現出任何擴張表型,然而,當同時缺失Cox10時,Dele1消融加重了Cox10?/?小鼠的表型,這些小鼠在約4周齡時死亡(圖4A)。與Cox10?/?小鼠相比,Cox10?/? Dele1?/?小鼠的體重進一步降低,心臟/體重比進一步增加,且心臟進一步擴張(圖4B-D),但不影響COX活性(圖4E)。在缺少Cox10的情況下,Dele1的缺失完全消除了Atf4和peIF2α的積累(圖4F-G),降低了ISR靶基因的表達(圖4H)。這些結果表明,Dele1的缺失與Oma1缺失類似同樣影響心臟特異性Cox10?/?小鼠的表型。ISR在Cox10?/?小鼠心臟中沿Oma1-Dele1軸被激活(圖4I),這與小鼠壽命延長有關。
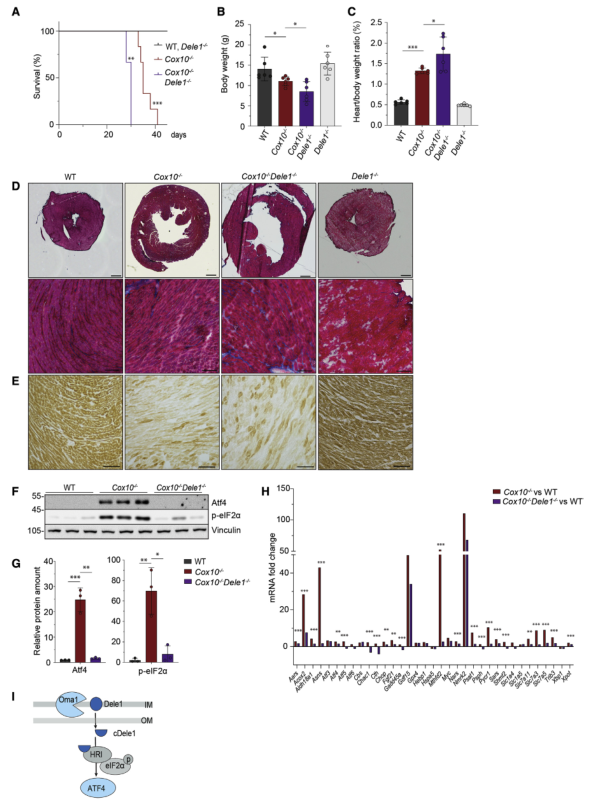
圖4 Cox10?/?小鼠心臟Dele1缺失惡化線粒體心肌病并損傷ISR
5、Oma1介導的ISR反應支持心臟的谷胱甘肽代謝
為確定OXPHOS缺乏時ISR受損的代謝后果,在4種小鼠心臟中進行靶向代謝組學研究(圖5A-B)。觀察到Cox10?/?心臟中單碳代謝中間體如絲氨酸、甘氨酸和蛋氨酸的積累,以及天冬酰胺、丙氨酸、支鏈氨基酸和脯氨酸的積累,Oma1的缺失沒有影響這些代謝物的穩態水平,也沒有強烈影響它們在Cox10?/?心臟中的積累(圖5A)。完全相反,Cox10?/?心臟中GSH的還原形式的積累在Cox10?/?Oma1?/?心臟中顯著增加(圖5B),這表明Oma1在OXPHOS缺乏時調節GSH水平。GSH代謝與單碳代謝密切相關,由Atf4和Nrf2共同調控(圖5C)。與Oma1缺失時ISR受損一致,Cox10?/?心臟中參與GSH代謝的酶的表達增加,而與Cox10?/?心臟相比,Cox10?/?Oma1?/?心臟中幾種酶的表達顯著降低(圖5D)。蛋白質組顯示,許多這些酶在 Cox10 缺陷的心臟中的積累和在沒有Oma1的情況下有較低的穩態水平(圖5E)。這些蛋白質包括GSH依賴的脂質過氧化物酶Gpx4,GSH降解酶Chac1和Chop1,它們的積累通過對心臟組織的WB進行監測(圖5F-G)。總之,得出結論,Oma1介導的ISR支持Cox10?/?心臟中的GSH代謝。
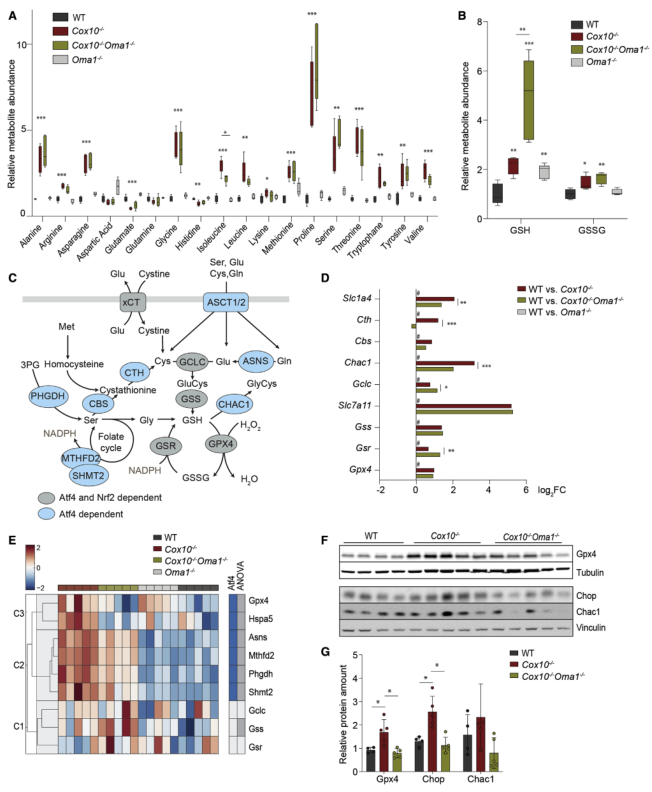
圖5 Oma1介導的ISR支持線粒體心肌病的谷胱甘肽代謝
6、Cox10?/?Oma1?/?心臟具有鐵死亡特征
對心臟RNA-seq數據中與細胞死亡相關的基因進行無監督聚類,發現這些基因在Cox10?/?和Cox10?/?Oma1?/?心臟中有不同的調控,包括促凋亡蛋白Chop和Chac1(圖6A)。心臟切片的TUNEL染色檢測到Cox10?/?中TUNEL陽性心肌細胞比Cox10?/?Oma1?/?中更多(圖6B-C)。然而,檢測到的TUNEL陽性細胞數量較少不能解釋Cox10?/?小鼠心肌病的嚴重程度,也不可能解釋Cox10?/?Oma1?/?小鼠的加重表型。此外,在Cox10?/?Oma1?/?心臟中存在炎癥反應(圖6D)。結合該結果以及GSH和鐵死亡有關等,因而檢測了MDA的積累。結果發現在Cox10?/?中能檢測到MDA的積累但是在Cox10?/?Oma1?/?心臟中積累顯著增加(圖6E,H)。透射電子顯微鏡觀察到脂質過氧化旋渦的形成(圖6F)。此外,染色也觀察到Cox10?/?Dele1?/?小鼠心臟中脂質過氧化的形成。證實這種死亡與鐵死亡有關。此外,與Cox10?/?Oma1?/?小鼠相似(圖5E-5G),Cox10?/?心臟中Dele1的丟失使Gpx4蛋白水平降低到野生型水平(圖6J)。因此,Oma1或Dele1的缺失,消除 Cox10-/-心臟中的ISR,與Gpx4蛋白水平降低和脂質過氧化增加有關,表明 Oma1-Dele1-Atf4 信號軸可防止OXPHOS缺陷心臟中的鐵死亡(圖6K)。
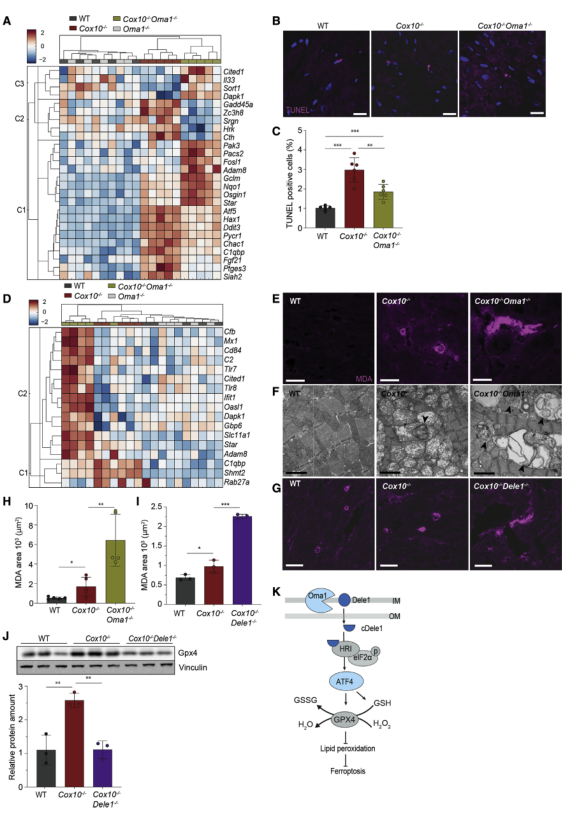
圖6 Cox10?/?小鼠心臟表現出Oma1依賴的鐵死亡特征和炎癥
7、依賴于Oma1的ISR在體外抵御鐵死亡
為明確ISR信號對鐵死亡的抵抗作用,分離4種小鼠的MEFs細胞。觀察到與WT、Cox10?/?、Oma1?/?細胞比較,Cox10?/?Oma1?/?細胞的存活率顯著下降(圖7A-B),但該情況在鐵死亡抑制劑Fer-1存在的情況下顯著抑制(圖7C),在鐵死亡誘導劑erastin存在的情況下加劇(圖7D-E)。提示Oma1保護Cox10?/?MEFs細胞抵御鐵死亡。
與體內結果類似,觀察到Atf4,GPX4,Chop在Cox10?/?而非Cox10?/?Oma1?/? MEFs細胞中積累(圖7F),同時,Atf4靶基因的Oma1依賴性表達增加,包括參與GSH代謝的各種酶(圖7G)。這些基因在Cox10?/?積累也依賴于Dele1(圖7H)。對這4種MEFs細胞的代謝組分析揭示了半胱氨酸、胱硫氨酸和谷胱甘肽在Cox10?/?而非Cox10?/?Oma1?/? MEFs細胞中積累(圖7I)。對Cox10?/?細胞施以eIF2α磷酸化和ISR信號通路抑制劑ISRIB處理。短時間用ISRIB處理增加野生型細胞對erastin的敏感性,而Cox10?/?的存活在這些條件下不受影響,在ISRIB長時間抑制ISR后,erastin誘導的Cox10?/?細胞鐵死亡增加(圖7J)。同樣,Atf4或Dele1缺失增加了Cox10?/?細胞對erastin誘導的鐵死亡的易感性(圖7K)。這些證明Oma1-Dele1介導的ISR對鐵死亡有抵御作用。
此外,圖7L-M進一步證明Oma1支持細胞對硒的利用并促進Gpx4的積累。因此,Oma1介導的ISR增加了轉硫作用途徑酶的轉錄,從而支持硒的利用,從而促進Gpx4的積累和對脂質過氧化和鐵死亡的抵抗力(圖7N)。
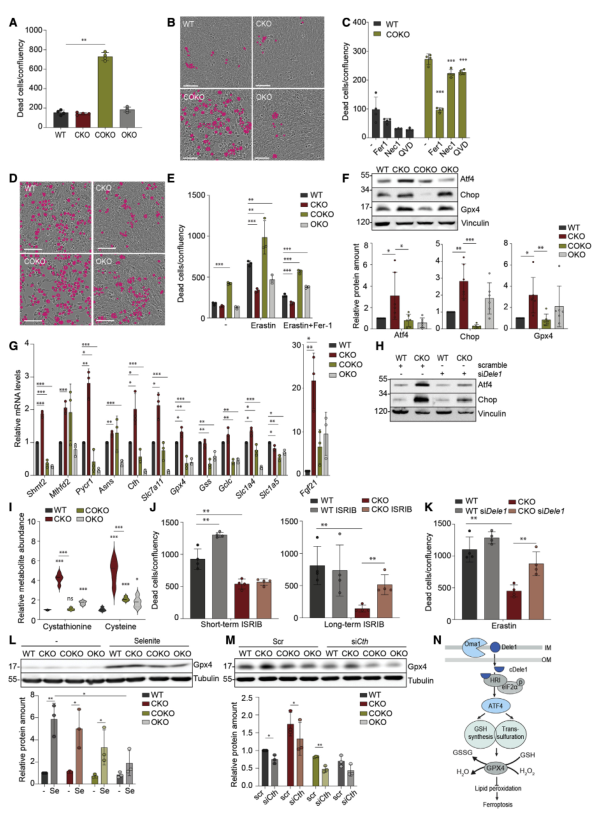
圖7 ISR沿Oma1-Dele1-Atf4軸激活保護Cox10?/? MEFs抵抗鐵死亡
總之,如圖8所示,Cox10?/?心臟中的 OXPHOS 缺乏會導致心肌病并引發沿 Oma1-Dele1-Atf4 軸的綜合應激反應。Oma1依賴的應激信號可保護谷胱甘肽代謝和Gpx4積累,以限制脂質過氧化、抑制鐵死亡和延緩心肌病。

圖8 圖形摘要
參考文獻:
Ahola Sofia., Rivera Mejías Pablo., Hermans Steffen., Chandragiri Srikanth., Giavalisco Patrick., Nolte Hendrik., Langer Thomas.(2022). OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab, undefined (undefined), undefined. doi:10.1016/j.cmet.2022.08.017