






姜黃烯醇通過lncRNA H19/miR-19b-3p/FTH1軸觸發肺癌細胞鐵死亡
肺癌是最常見的癌癥類型之一,是全球癌癥相關死亡的主要原因。盡管現有藥物在晚期肺癌的治療中取得了許多進展,但由于其相關的嚴重不良反應、劑量限制性毒性、耐藥和較差的選擇性,其治療效果有限。因此,迫切需要開發新的治療策略或新的有效藥物來治療肺癌。姜黃烯醇是溫瘀金的有效成分,已被報道在幾種腫瘤中發揮其抗腫瘤潛力。然而,姜黃烯醇在肺癌中的作用和分子機制尚不清楚。該研究發表于《Bioactive Materials》,IF:12.91。
技術路線:

主要研究結果:
1. 姜黃素誘導肺癌細胞死亡,抑制肺癌細胞增殖
為了評估姜黃烯醇對肺癌的抗癌潛力,研究者在人肺成纖維細胞(CCD19),人正常肺上皮細胞(BEAS-2B)和肺癌細胞(H1299和H460)中使用不同濃度的姜黃烯醇處理24小時。研究者發現,姜黃烯醇在肺癌細胞中以濃度依賴性方式抑制細胞存活,但在正常肺細胞中毒性相對較小(圖1A和B)。表明莪術醇對肺癌細胞有選擇性的細胞毒性。克隆形成實驗檢測姜黃烯醇對細胞增殖的影響;如圖1C和D所示,與對照組相比,莪術醇以劑量依賴性方式顯著抑制肺癌細胞的集落形成。采用Annexin V-FITC/PI雙染流式細胞術檢測姜黃烯醇對H1299和H460細胞死亡的影響。與對照組相比,姜黃烯醇處理后的肺癌細胞中死亡細胞的百分比較高。然而,總體的細胞凋亡較低(圖1E和F),這可能是由于凋亡不是姜黃烯醇誘導的細胞死亡的主要模式。綜上所述,這些數據表明姜黃烯醇降低肺癌細胞的活力并誘導細胞死亡。

圖1 姜黃烯醇處理H1299和H460細胞后檢測細胞活力
2. 鐵死亡是姜黃素誘導肺癌細胞死亡的主要方式
采用程序性壞死抑制劑Nec-1、自噬抑制劑氯喹、泛caspase抑制劑Z-VAD以及鐵死亡抑制劑去鐵胺(DFO)、Ferrostatin (Fer-1)和liprostatin -1 (Lip-1)評估姜黃烯醇誘導的主要細胞死亡程序。結果表明,Nec-1, Z-VAD和CQ不能明顯抑制姜黃烯醇在H1299和H460細胞中引起的細胞死亡(圖2A-C)。然而,鐵死亡抑制劑DFO、Fer-1和Lip-1顯著挽救了姜黃烯醇誘導的細胞死亡(圖2D-F)。這些結果證明,鐵死亡可能是姜黃烯醇誘導的主要細胞死亡程序。
為進一步確定姜黃烯醇是否通過鐵死亡誘導細胞死亡,western blot檢測鐵死亡相關蛋白。研究者的數據顯示姜黃烯醇處理顯著增加了肺癌細胞中血紅素加氧酶1 (HO-1)和轉鐵蛋白的表達水平,但降低了谷胱甘肽過氧化物酶4 (GPX4)、溶質載體家族40成員1 (SLC40A1)、溶質載體家族7成員11 (SLC7A11)、鐵蛋白重鏈1 (FTH1)、核因子e2相關因子2 (NRF2)和谷氨酰胺酶的表達水平(圖3A)。此外,鐵螯合劑DFO可以挽救姜黃烯醇誘導的鐵死亡蛋白的表達(圖3B)。
在隨后的實驗中,研究者進行了RNA測序,以研究姜黃烯醇處理后的差異轉錄組反應。采用數字基因表達譜技術篩選并鑒定差異表達基因。進一步對差異表達基因進行基因本體論(GO)分類和京都基因與基因組百科全書(KEGG)通路分析。因此,在H1299細胞中確定了1,665個上調基因和1,232個下調基因。在H460細胞中確定了1,484個上調基因和759個下調基因。GO富集分析顯示,氧化還原過程和氧化還原酶活性得到富集。同時,KEGG分析發現鐵死亡通路被富集(圖3C和D)。這些結果表明姜黃烯醇處理后肺癌細胞發生了鐵死亡。
眾所周知,活性氧(ROS)蓄積、還原型谷胱甘肽(GSH)耗竭、脂質過氧化和氧化還原活性鐵過載是鐵死亡過程中的關鍵事件。檢測細胞內活性氧(ROS)、還原型谷胱甘肽(GSH)、氧化應激標志物丙二醛(MDA)和細胞內螯合鐵水平。正如預期的那樣,姜黃烯醇處理后觀察到ROS水平增加(圖4A)和GSH水平降低(圖4B和C)。此外,鐵螯合劑DFO可以消除姜黃烯醇引起的GSH耗竭(圖4D和E)。同時,姜黃烯醇處理上調了肺癌細胞中的MDA水平(圖4F)。此外,鐵敏感熒光團Phen Green SK被用來監測細胞內螯合鐵,鐵結合后被淬滅。正如預期的那樣,與對照組相比,姜黃烯醇處理降低了Phen Green sk陽性細胞的比例,而鐵螯合劑DFO可以消除這一現象(圖4G),表明鐵死亡被觸發。因此,這些結果表明鐵死亡是姜黃烯醇觸發肺癌細胞死亡的主要機制。

圖2 姜黃烯醇單獨或聯合不同細胞死亡抑制劑對肺癌細胞活力的影響

圖3 姜黃烯醇誘導肺癌細胞發生鐵死亡
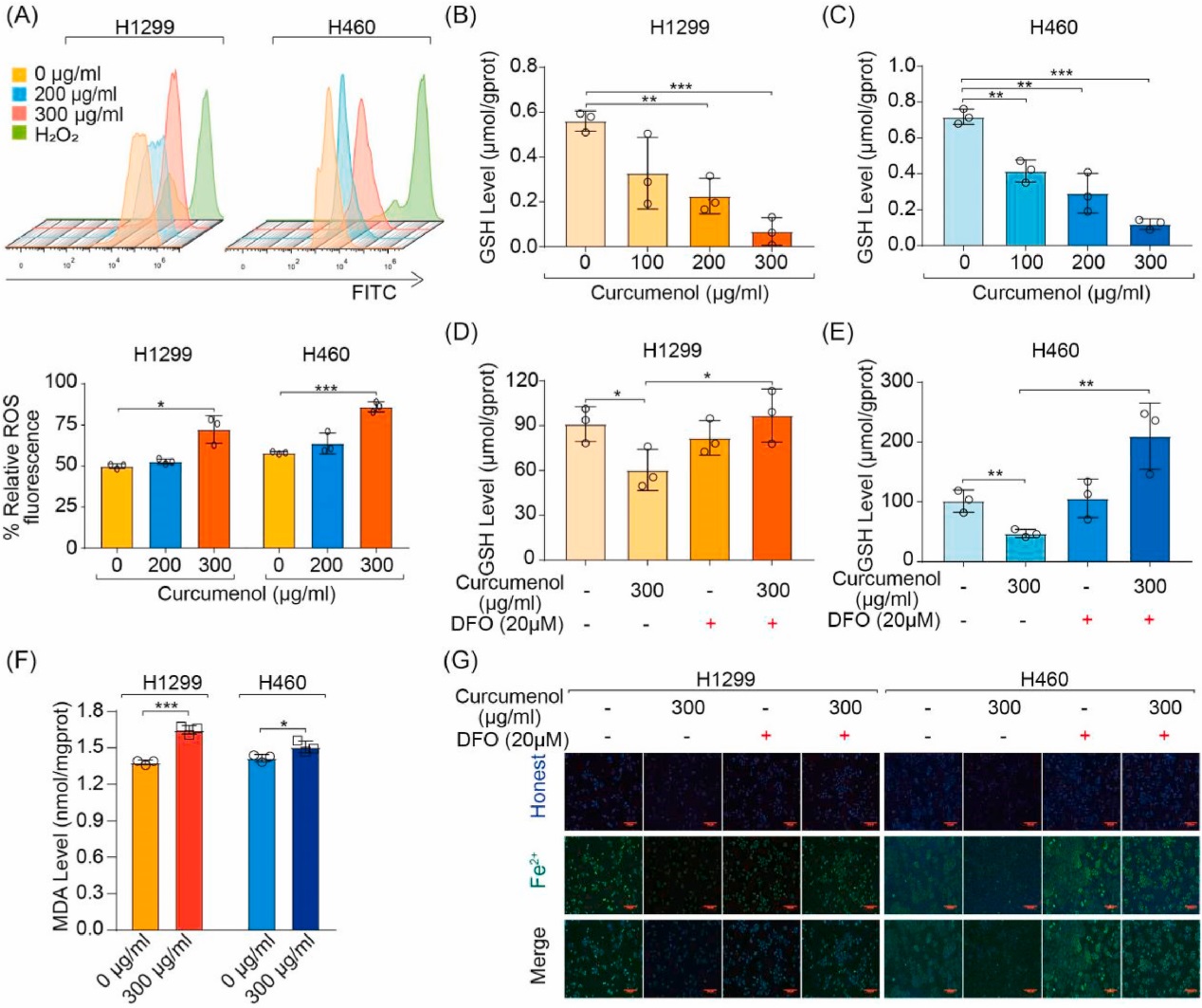
圖4 鐵死亡促進了姜黃烯醇誘導的肺癌細胞死亡
3. 姜黃烯醇在肺癌異種移植模型中引發鐵死亡
建立肺癌皮下移植瘤模型,評價莪術醇的體內抗腫瘤作用。皮下移植瘤體積達80 ~ 100 mm3后,將裸鼠隨機分為溶劑組、莪術醇組(200 mg/kg/d)、去鐵胺組(100 mg/kg/d)和藥物聯合組。研究者的數據顯示,姜黃烯醇顯著抑制了異種移植瘤的生長,并且姜黃烯醇的抗腫瘤作用可以被鐵死亡抑制劑DFO消除(圖5A和B)。然而,對照組和姜黃烯醇處理組的體重沒有統計學差異(圖5C),表明姜黃烯醇沒有明顯的毒性。在進一步的研究中,研究者通過免疫組織化學(IHC)研究了姜黃烯醇對鐵死亡的影響。姜黃烯醇處理后,GPX4, FTH1和SLC7A11的水平降低,HO-1和轉鐵蛋白的表達增加,表明姜黃烯醇觸發鐵死亡。此外,與鐵螯合劑DFO共處理后,姜黃烯醇誘導的細胞死亡幾乎被消除(圖5D)。綜上所述,研究者的結果表明姜黃烯醇在體內誘導肺癌細胞發生鐵死亡。
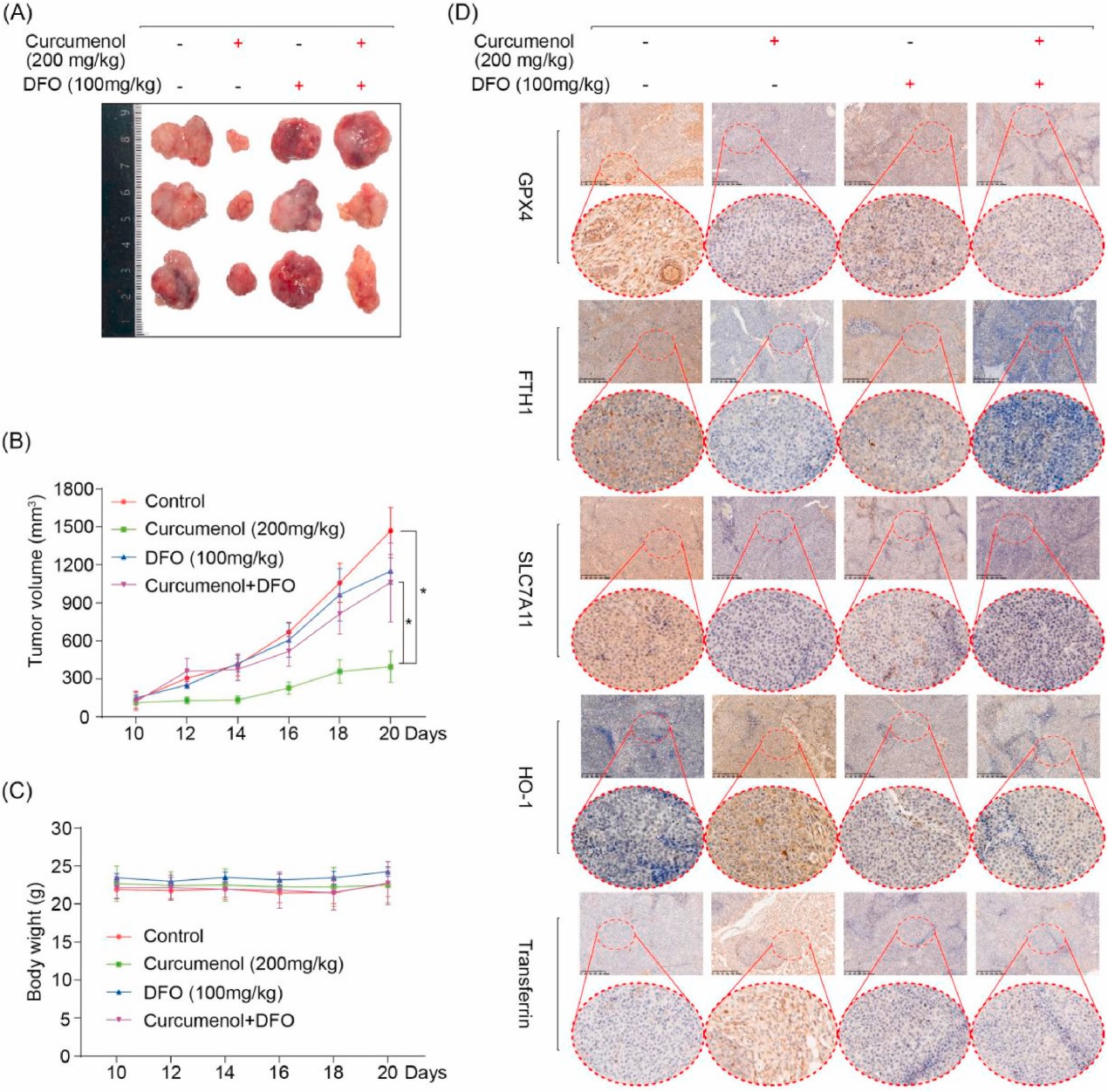
圖5 姜黃烯醇在肺癌異種移植模型中觸發鐵死亡
4. lncRNA H19表達與姜黃烯醇誘導鐵死亡的相關性
為了確定lncRNA在姜黃烯醇抗癌作用中的作用,研究者進行了RNA測序。研究者發現,與未處理的細胞相比,姜黃烯醇處理的肺癌細胞中lncRNA H19顯著下調(圖6A)。類似的結果通過qRT-PCR進一步證實(圖6B)。為了研究lncRNA H19在姜黃烯醇誘導的鐵死亡中的生物學功能,研究者在H1299和H460細胞中分別通過轉染過表達質粒和shRNA來過表達或敲低lncRNA H19。lncRNA H19過表達顯著增加了鐵死亡負調控因子(NRF2、GPX4、FTH1和SLC7A11)的表達水平(圖6C-E)。相比之下,lncRNA H19敲低顯著降低了鐵死亡負調控因子(NRF2、GPX4、FTH1和SLC7A11)的表達水平(圖6F-H)。因此,研究者的結果表明lncRNA H19參與了姜黃烯醇誘導的鐵死亡。
為了研究lncRNA H19對姜黃烯醇誘導的鐵死亡的影響,研究者在H1299和H460細胞中轉染過表達質粒或shRNA。克隆形成實驗和CCK-8結果顯示,lncRNA H19過表達顯著消除了姜黃烯醇對H1299和H460細胞的抑制作用(圖7A-F)。相反,lncRNA H19敲低顯著抑制了H1299和H460細胞的克隆形成,增強了姜黃烯醇對這些細胞的抗癌作用(圖7G-L)。綜上所述,這些數據表明lncRNA H19是姜黃烯醇誘導鐵死亡的關鍵決定因素。

圖6 lncRNA H19表達與姜黃烯醇誘導鐵死亡的相關性
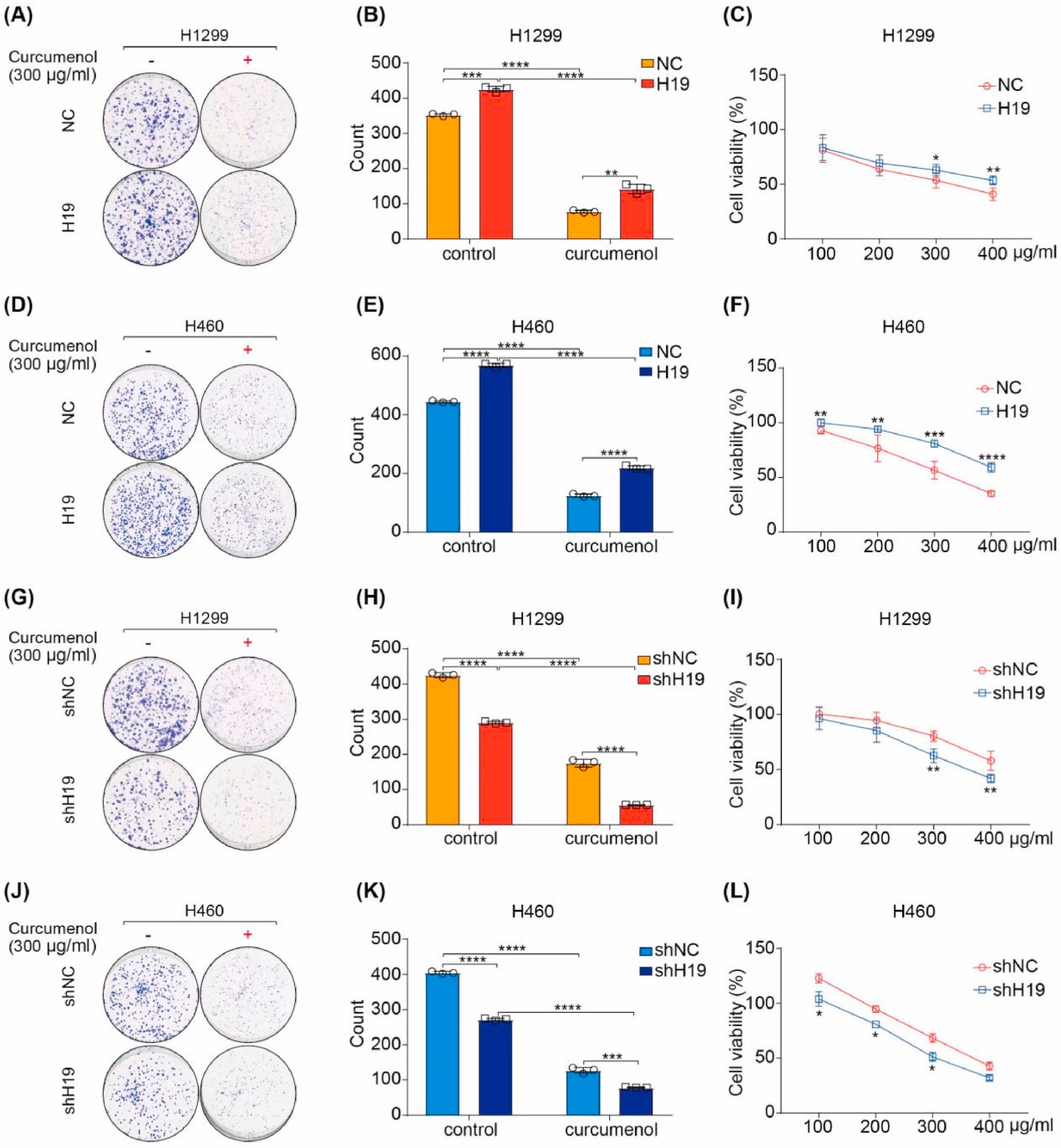
圖7 lncRNA H19對姜黃烯醇誘導鐵死亡的影響
5. 姜黃素通過lncRNA H19/miR-19b-3p/FTH1軸誘導肺癌細胞鐵死亡
越來越多的證據表明,lncRNA可以作為ceRNA或與RNA結合蛋白相互作用來調控靶基因的表達。因此,研究者基于生物信息學數據庫(Starbase)中lncRNA- miRNA和miRNA-mRNA的相互作用構建了ceRNA網絡,發現lncRNA H19可能靶向miR-181a-5p、miR-19b-3p、miR-200b和miR-675。在4個候選miRNA中,與對照組相比,經莪術醇處理后,H1299和H460細胞中miRNA-19b-3p的表達水平顯著升高(圖8A-C)。有趣的是,在泛癌分析中,miR-19b-3p與FTH1在肺鱗狀細胞癌(圖8D)和腺癌中呈負相關。由于研究者已經證明lncRNA H19可以調控H1299和H460細胞中FTH1的表達(圖6C-H),研究者有理由推測lncRNA H19通過靶向miR-19b-3p調控FTH1水平。結果顯示,lncRNA H19過表達抑制H1299和H460細胞中miR-19b-3p水平(圖8E)。相比之下,lncRNA H19敲低增強了H1299和H460細胞中miR-19b-3p的表達(圖8F)。同時,研究者發現miR-19b-3p mimics降低了FTH1的表達,但增加了姜黃烯醇在H1299和H460細胞中的抗癌活性(圖8G-L)。相反,miR-19b-3p抑制劑增加了FTH1的表達,但減弱了姜黃烯醇在H1299和H460細胞中的抗癌活性(圖8M-R)。這些數據表明姜黃烯醇通過lncRNA H19/miR-19b-3p/FTH1軸觸發肺癌細胞的鐵死亡。
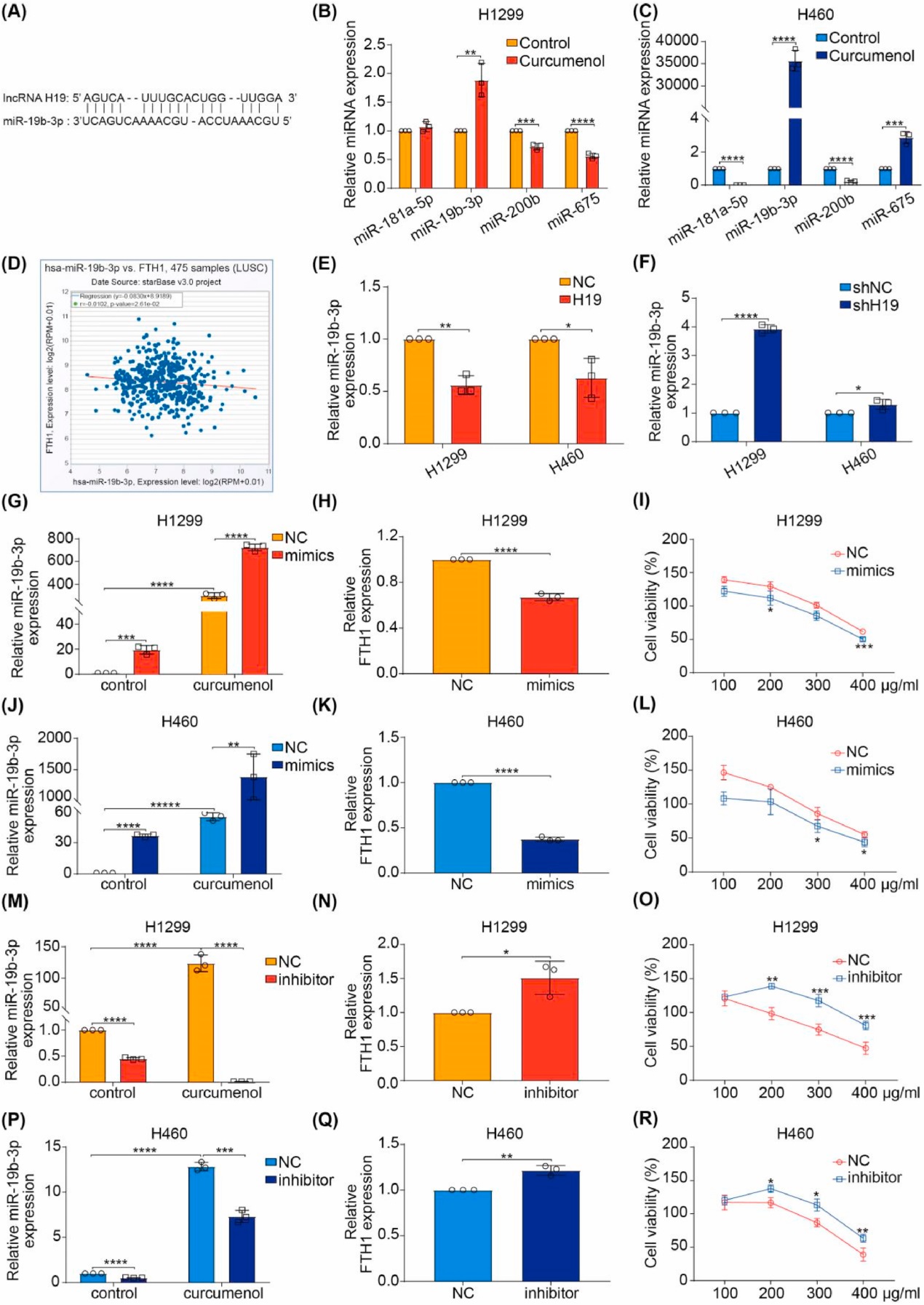
圖8 姜黃烯醇通過lncRNA H19/miR-19b-3p/FTH1軸誘導肺癌細胞鐵死亡
結論:
該研究表明,天然產物姜黃烯醇通過觸發鐵死亡發揮其抗腫瘤作用,并且lncRNA H19/miR-19b-3p/FTH1軸在姜黃烯醇誘導的鐵死亡細胞中發揮重要作用。本研究有望為肺癌患者提供潛在的治療藥物。
參考文獻:
Ruonan Zhang, Ting Pan, Yu Xiang, et al. Curcumenol triggered ferroptosis in lung cancer cells via lncRNA H19/miR-19b-3p/FTH1 axis. Bioactive Materials. Volume 13, 2022, Pages 23-36, ISSN 2452-199X, https://doi.org/10.1016/j.bioactmat.2021.11.013.