






在胰管腺癌中,LncRNA-PACERR通過與miR-671-3p和m6A-reader IGF2BP2相互作用誘導腫瘤前巨噬細胞
胰腺導管腺癌(PDAC)是胰腺癌的主要類型,是最具侵襲性和致命性的消化道惡性腫瘤,預后極差。PDAC的嚴重預后與腫瘤微環境(TME)相關,其中組織相關的巨噬細胞(TAM)是PDAC TME中主要的免疫細胞。目前,有研究發現LncRNA-PACERR是PDAC TME中TAMs的關鍵調控因子,并揭示了其在細胞質和細胞核中的新機制。該研究于2022年5月7日發表在《Journal of Hematology & Oncology》,IF:17.388。
技術路線:

主要研究結果:
1. LncRNA-PACERR在TAMs中過表達,PACERR+ TAMs浸潤升高與PDAC患者預后不良相關
為了確定PDAC中TAMs中LncRNA-PACERR的表達水平,使用磁激活細胞分選(MACS)從46名PDAC患者的癌旁組織中分離出CD163+細胞和CD80+細胞,這些巨噬細胞的體積為0.5 cm3。通過流式細胞儀分析確保了分選細胞的高純度(圖1b)。對腫瘤組織和非腫瘤組織組成的PDAC組織微陣列(TMAs)的FISH免疫熒光分析表明,LncRNA-PACERR在TAMs中的表達明顯高于正常組織常駐巨噬細胞(M1-NTRMs) (圖1c,d),并且CD163和LncRNA-PACERR之間存在很強的共定位關系(圖1e)。Kaplan-Meier分析顯示LncRNA-PACERR+ TAMs高浸潤與PDAC預后不良相關 (圖1f)。綜上所述,這些結果表明LncRNA-PACERR+ TAMs越多,PDAC預后越差。
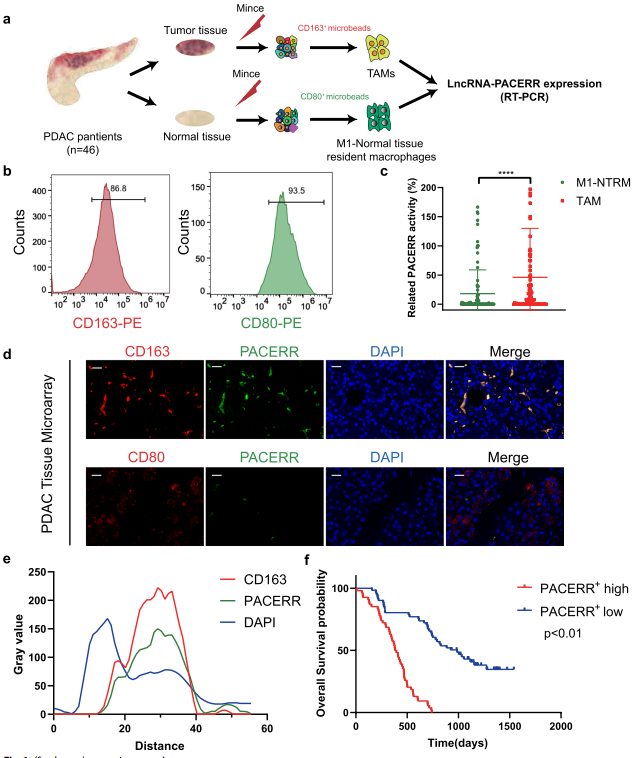
圖1 TAMs中LncRNA-PACERR表達在PDAC組織中被激活,與預后不良相關
2. LncRNA-PACERR+ TAMs具有M2極化巨噬細胞的特征,在體外和體內均能促進胰腺癌細胞的增殖、遷移和侵襲
為了明確PACERR的功能,利用THP-1細胞系和PATU-8988/PANC-1細胞構建TAMs體外共培養模型(圖2a)。qPCR分析證實,TAMs敲除LncRNA-PACERR后,M2巨噬細胞標志物CD206、CD163、Arginase-1、TGF-β、IL-10、IL-6表達水平下調(圖2a)。流式細胞術顯示,THP-1來源的TAMs中LncRNA-PACERR表達減少,導致CD206+和CD163+巨噬細胞比例降低(圖2b)。基于克隆形成和CCK8實驗,TAMs中LncRNA-PACERR敲低可抑制胰腺癌細胞增殖(圖2c)。Transwell實驗表明TAMs中LncRNA-PACERR增加了胰腺癌細胞的遷移和侵襲能力(圖2d, e)。用兩組小鼠來展示小鼠的預后,觀察到LncRNA-PACERR敲低組小鼠的總生存期較陰性對照組小鼠更長(圖3a-e)。對于轉移小鼠模型,其皮下腫瘤模型和轉移性肝細胞數量減少,CD163+細胞的數量和CD206 +細胞轉移焦點在TAMs中LncRNA-PACERR敲除后降低(圖3h-f)。
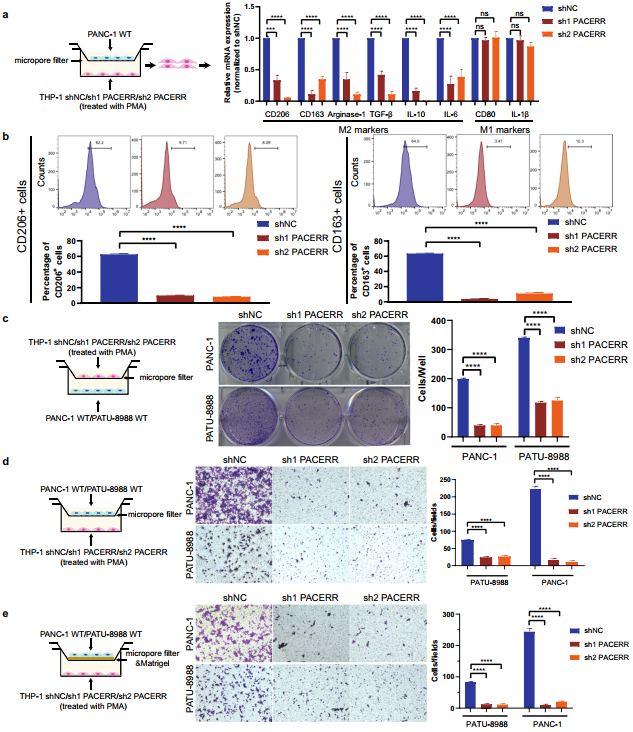
圖2 LncRNA-PACERR的敲低在體外抑制THP -1衍生的TAMs的M2極化和促腫瘤功能
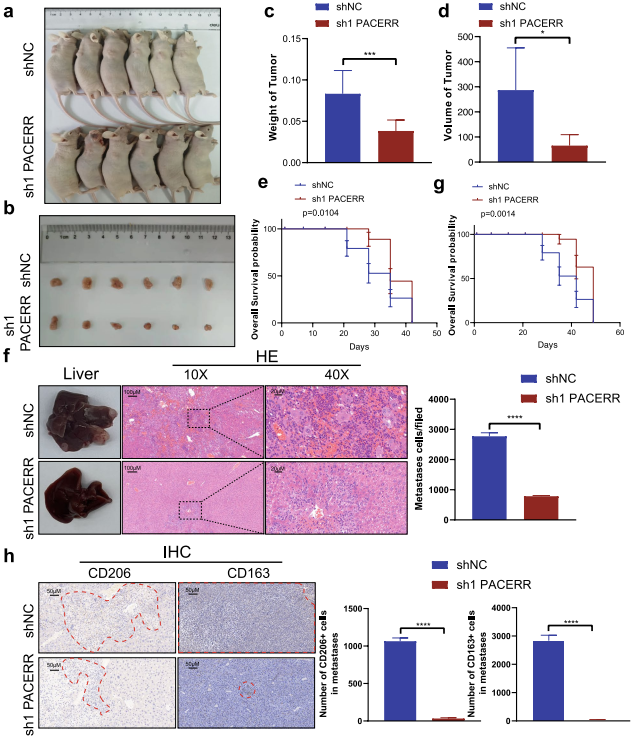
圖3在體內LncRNA-PACERR+ TAMs促進PDAC細胞生長和肝轉移
3. LncRNA-PACERR通過吸附miR-671-3p上調KLF12/p-AKT/c-myc軸
LncRNA-PACERR促進惡性腫瘤進展的分子機制:通過FISH和細胞分離實驗確定了LncRNA-PACERR的亞細胞位置,揭示了LncRNA-PACERR在TAMs細胞質和細胞核中的主要分布(圖4a-d)。qPCR檢測了兩個假定的LncRNA-PACERR靶標miRNA (miR-4448和miR-671-3p) (圖4e)。在THP-1衍生的TAMs中,敲低LncRNA-PACERR后,miR-671-3p上調,而miR-4448未發生變化(圖4e)。因此選擇miR-671-3p進行進一步研究。作者發現LncRNA-PACERR對pri-miRNA或pre-miRNA的表達或啟動子活性沒有顯著影響(圖4f-h)。RIP結果顯示,AGO2顯著結合LncRNA-PACERR和miR-671-3p(圖4i)。雙熒光素酶報告基因檢測確定了miR-671-3p與LncRNA-PACERR的相互作用(圖4j,k)。此外,利用臨床樣本檢測LncRNA-PACERR和miR-671-3p的表達,發現miR-671-3p在TAMs中的表達低于M1-NTRMs,并且與LncRNA-PACERR呈負相關(圖4i, m)。為評估miR-671-3p的臨床預后,對PDAC TMAs的患者進行分組,分為miR-671-3p+高組和miR-671-3p+低組(圖4n)。Kaplan-Meier分析顯示,miR-671-3p表達越高,PDAC的預后越好(圖4n)。另外,作者對PDAC中報道過的最著名的靶基因KLF12,進行了確認(圖5a)。qPCR分析和IF分析證實,KLF12在TAMs中高表達,與不良預后相關(圖5b)。為了探究miR-671-3p是否可以直接海綿化KLF12,首先測定PDAC樣本中miR-671-3p的RNA表達水平,發現KLF12與miR-671-3p之間存在負相關 (圖5c)。此外,在THP-1衍生的TAMs中,轉染miR-671-3p模擬物后KLF12 mRNA表達水平降低,轉染miR-671-3p抑制劑后KLF12 mRNA表達水平升高(圖5d)。雙熒光素酶報告基因檢測顯示,用miR-671-3p模擬物轉染HEK-293 T細胞后,Luc-KLF12-wt組的熒光素酶活性降低,而Luc-KLF12-mut組的熒光素酶活性沒有變化(圖5e, f)。此外,在TAMs中,KLF12的表達與LncRNA-PACERR的表達水平呈正相關(圖5g)。轉染miR-671-3p抑制劑后,LncRNA-PACERR敲低,可挽救THP-1中KLF12的表達(圖5h, i)。轉染LncRNA-PACERR過表達質粒增加了Luc-KLF12-wt的熒光素酶活性,轉染LncRNA-PACERR和miR-671-3p模擬物減弱了HEK-293 T的這種作用(圖5j)。western blot分析顯示LncRNA-PACERR調控KLF12顯著促進AKT磷酸化,通過下調THP-1衍生的TAMs中的PTEN增加c-myc的表達(圖5k, l)。qPCR、流式細胞術、CCK8、集散形成和Transwell實驗得到與LncRNA PACERR相似的結果,并且過表達KLF12可以促進LncRNA-PACERR的表達(圖5m)。綜上所述,LncRNA-PACERR可以通過隔離miR-671-3p來釋放KLF12,從而通過KLF12/AKT/c-myc通路實現TAMs促進胰腺癌惡性進展。
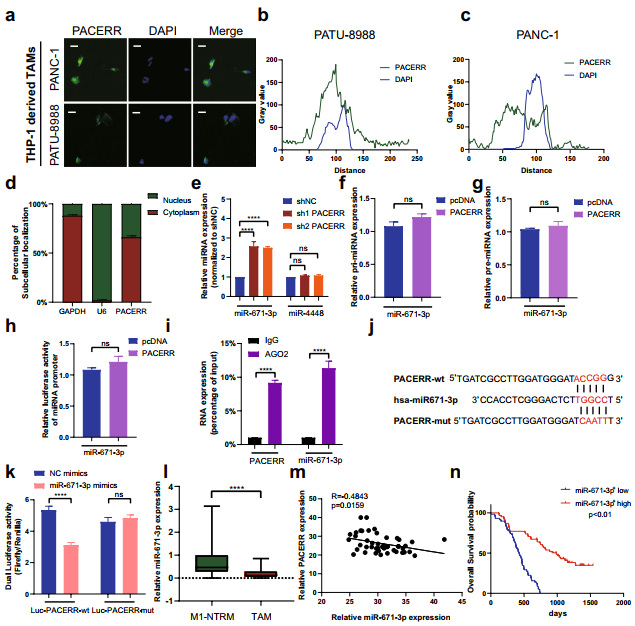
圖4 LncRNA-PACERR在TAMs中作為ceRNA海綿作用于miR-671-3p
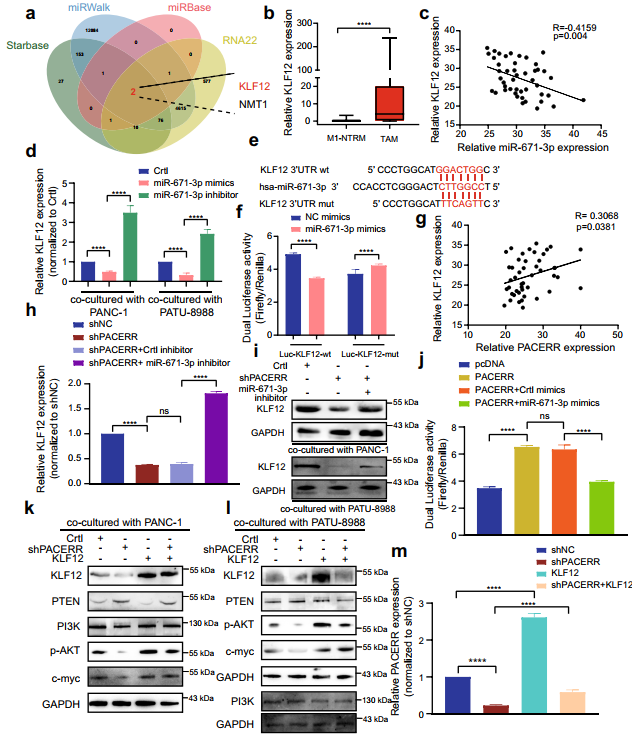
圖5 MiR-671-3p直接結合KLF12的3'UTR,調控KLF12/AKT/c-myc軸
4. KLF12/LncRNA-PACERR復合物通過招募EP300來促進LncRNA - PACERR在細胞核內的轉錄
根據圖5m推測KLF12可能促進LncRNA-PACERR在細胞核內的轉錄。通過ChIP實驗確定KLF12與TAMs中LncRNA-PACERR啟動子區結合(圖6a)。根據KLF12的motif預測了LncRNA-PACERR啟動子區域的結合位點,發現KLF12結合在LncRNA-PACERR上游的-302—+ 100bp處(圖6b-d)。鑒于大多數轉錄因子通過影響組蛋白修飾來調控基因轉錄,因此通過ChIP分析確定KLF12在TAMs中LncRNA-PACERR的TSS處增加了H3K27ac的富集(圖6f)。因此,co-IP實驗驗證了KLF12會招募組蛋白乙酰轉移酶EP300 (圖6g)。RNA-pull down和RIP實驗驗證KLF12會與細胞核內的LncRNA-PACERR結合 (圖6h, i)。綜上所述,這些數據表明LncRNA-PACERR也直接受到KLF12的轉錄調控。
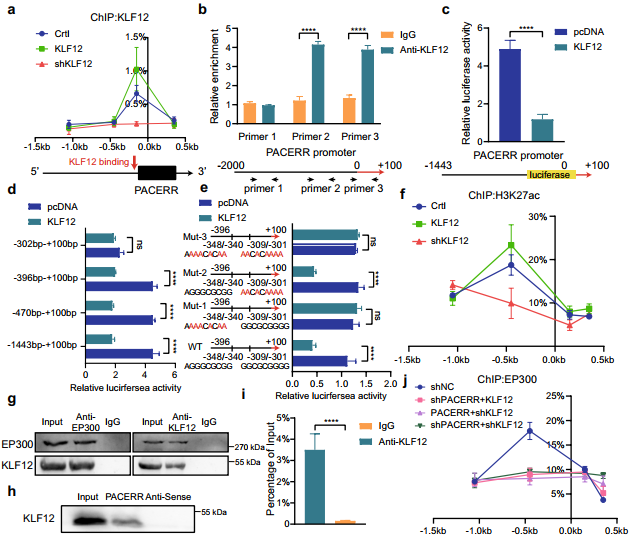
圖6 KLF12直接與LncRNA-PACERR結合,并以LncRNA-PACERR依賴的方式將EP300招募到LncRNA-PACERR啟動子區域
5. LncRNA-PACERR在TAMs中通過與IGF2BP2協同發揮致癌作用
首先通過RNA-pull down和質譜分析來篩選出于LncRNA-PACERR相互作用的m6a相關蛋白-IGF2BP2(圖7a)。然后,進一步驗證了IGF2BP2作為LncRNA-PACERR的RNA結合蛋白(圖7b)。另外,RIP實驗也證實了IGF2BP2與LncRNA-PACERR之間的關系(圖7c)。此外,THP-1驅動的TAMs中LncRNA-PACERR FISH和IGF2BP2的共定位分析支持了它們的相互作用(圖7d)。RNA pull-down和western blot分析確定LncRNA-PACERR的1-293nt區域與IGF2BP2相互作用(圖7e, f)。最后,構建了含有標記截短和全長IGF2BP2的HEK-293 T細胞,并通過RIPqPCR檢測發現,KH1和KH2結構域對LncRNA-PACERR的招募具有決定性作用(圖7g,h)。而qPCR、western blot、FISH和IF檢測,LncRNA-PACERR和IGF2BP2彼此的表達水平并不改變(圖7d, j, k)。這些結果表明LncRNA-PACERR與IGF2BP2存在物理結合作用。
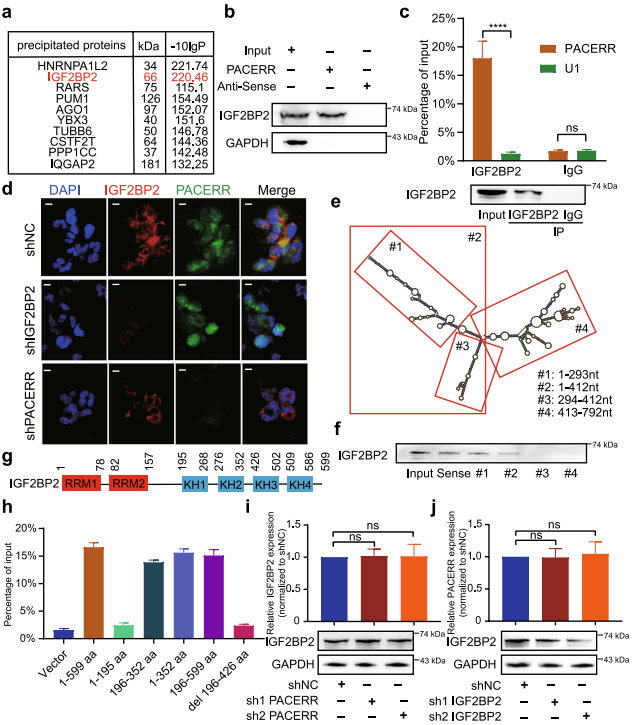
圖7 TAMs中LncRNA-PACERR直接與IGF2BP2結合
6. LncRNA-PACERR通過與IGF2BP2協同提高KLF12和c-myc的mRNA穩定性
由于KLF12和c-myc作為LncRNA-PACERR的下游靶點,推測LncRNA-PACERR是否與IGF2BP2共同調控KLF12和c-myc的穩定性。為了證實這一假設,首先使用qPCR和western blot分析來闡明LncRNA-PACERR/IGF2BP2對KLF12和c-myc表達的正向調控作用(圖8a,b)。放線菌素D發現,IGF2BP2敲除消除了LncRNA-PACERR過表達增加的KLF12和c-myc的穩定。LncRNA-PACERR敲除通過IGF2BP2過表達解除了KLF12和c-myc的半衰期(圖8c-f)。為了進一步闡明KLF12和c-myc是否被m6A甲基化修飾,使用基于GGAC m6A核心基序的m6A RIP qRT-PCR分析表明,在甲基轉移酶樣14(METTL14)敲除THP-1細胞中,KLF12轉錄本3'UTR的m6A甲基化和c-myc轉錄本的編碼區不穩定決定簇(CRD)區域降低(圖8g)。為了探究LncRNA-PACERR是否影響IGF2BP2與KLF12和c-myc m6a修飾區域的結合,通過RIP-qPCR檢測,LncRNA-PACERR敲低顯著降低了IGF2BP2與KLF12和c-myc m6a修飾區域的結合(圖8h, i)。鑒于IGF2BP2調節的m6A修飾區域主要位于mRNA 3'UTR的起始點,因此構建KLF12 3'UTR的熒光素酶報告載體,然后通過RIP-qPCR驗證野生型報告基因中IGF2BP2和m6A比突變報告基因中富集(圖8j-l)。此外,雙熒光素酶報告分析顯示,LncRNA-PACERR增加了KLF12-3'UTR野生型報告基因中的熒光素酶活性,與IGF2BP2的結果相似(圖8m)。這些數據表明,LncRNA-PACERR與IGF2BP2合作,增強了KLF12和c-myc在TAMs中的穩定性。
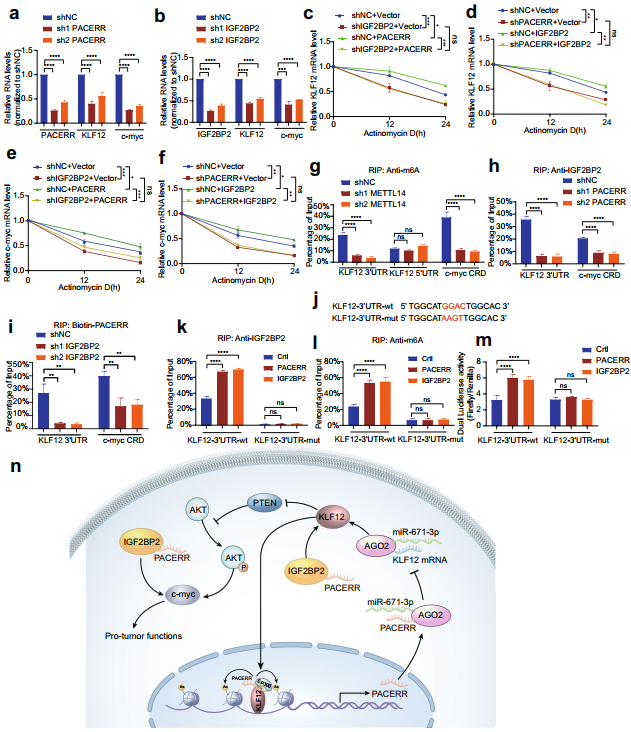
圖8 LncRNA-PACERR與IGF2BP2以m6a依賴的方式共同調控KLF12和c-myc
結論:
LncRNA-PACERR通過兩個途徑促進胰腺癌的惡性進展:作為ceRNA海綿體的miR-671-3p激活KLF12/ AKT/c-myc通路,并與IGF2BP2相互作用增強KLF12和c-myc的穩定性。同時,KLF12轉錄的LncRNA-PACERR與KLF12直接相互作用,KLF12/PACERR復合物通過招募EP300激活LncRNA-PACERR轉錄,從而促進TAMs中的M2極化和腫瘤前功能。