AlphaFold蛋白質結構數據庫
蛋白質是具有重要生物學功能的重要大分子,因此廣泛參與多項研究活動以及醫學和生物技術應用,從抗擊傳染病到應對環境污染 都發揮重要作用。了解蛋白質原子的三維排列可為理解蛋白質功能的作用和機制提供重要線索。然而,雖然通用蛋白質資源 (UniProt) 存檔了近 2.2 億個獨特的蛋白質序列,但蛋白質數據庫 (PDB) 僅保存了超過55000種不同蛋白質的180000多個3D結構,因此蛋白質3D結構解析嚴重限制了序列空間的覆蓋范圍支持全球生物分子研究。
用實驗確定的高分辨率結構實現對序列空間的更高覆蓋率是非常勞動密集型的。它通常需要大量的試驗和錯誤,例如,找到合適的構建體或蛋白質適合結晶的條件。盡管電子冷凍顯微鏡和用于結構確定的混合和綜合方法 (I/HM) 領域的最新進展加快了結構確定的步伐,但已知蛋白質序列與實驗蛋白質結構之間的差距仍在繼續擴大。縮小這一差距的一種方法是預測數百萬種蛋白質的結構。越來越多的研究人員部署人工智能 (AI) 技術,僅根據氨基酸序列計算預測蛋白質的結構。
AlphaFold 是由 DeepMind 開發的 AI 系統,可根據氨基酸序列對蛋白質結構進行最先進的預測。AlphaFold的準確性和速度允許創建一個大規模的結構預測數據庫。它將使生物學家能夠獲得幾乎任何蛋白質序列的結構模型,從而改變他們解決研究問題的方式并加速他們的項目。AlphaFold DB(https://alphafold.ebi.ac.uk)是基于AlphaFold算法構建的蛋白質3D機構預測數據庫。AlphaFold DB 的初始版本包含超過360000個預測結構、相應的元信息和置信度指標。預測目前涵蓋UniProt參考蛋白質組中16-2700 個氨基酸長度范圍內的大多數序列(以及覆蓋更長人類蛋白質的 1400 個殘基片段)。
AlphaFold DB 通過網頁提供對其預測的便捷訪問。這些頁面包含對 AlphaFold 系統的介紹,解決最常見的問題,允許批量下載完整的蛋白質組,并提供搜索引擎以查找特定于感興趣蛋白質的頁面。用戶可以通過基因名稱、蛋白質名稱、UniProt 登錄或生物名稱進行搜索。
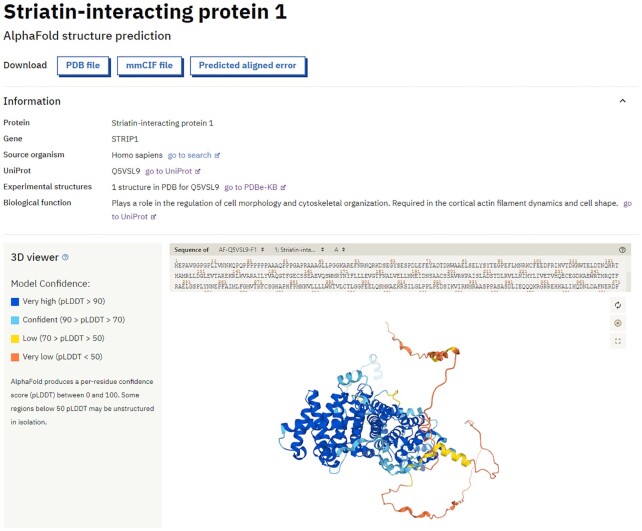
每個蛋白質都有一個專門的結構頁面,顯示基本信息(來自 UniProt和 PDBe)和 AlphaFold 模型的三個獨立輸出。前兩個輸出是3D坐標和每個殘基置信度度量 pLDDT,用于在集成的3D分子查看器 Mol中對模型的殘基進行著色。模型置信度可能會沿著一條鏈發生顯著變化,因此在解釋結構特征之前分析置信度度量是必不可少的。
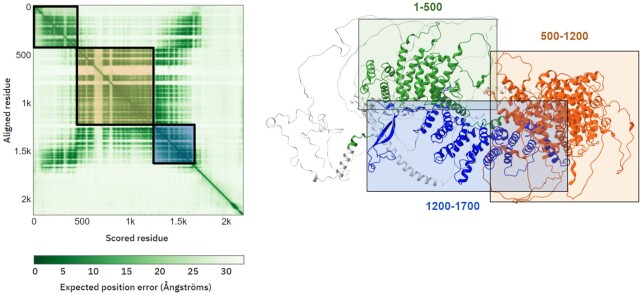
第三個輸出是成對置信度預測,它有助于評估相對域位置和方向的可靠性以及蛋白質的全局拓撲結構。該圖由成對的 PAE 值著色,它可以幫助用戶識別哪些域具有可靠地預測的相對于彼此的位置和方向,其中深綠色表示高置信度。在繪圖中選擇一個區域也會在 3D 查看器中突出顯示序列的相應部分。