






在KEAP1缺失的肺癌中,靶向CoQ-FSP1軸驅動鐵死亡和輻射抗性
鐵死亡是脂質過氧化引起的一種特殊的細胞死亡方式,在癌癥治療中,由于對特定癌癥遺傳背景下的鐵死亡機制的不完全理解而受到阻礙。KEAP1在肺癌中經常發生突變或失活,而KEAP1突變型肺癌對包括放療在內的大多數治療方法均有難治性。在本研究中,作者探討了鐵死亡抑制蛋白1 (FSP1,也被稱為AIFM2)在肺癌中的作用機制。本文于2022年4月發表于《Nature communications》, IF=12.121。
本文技術路線:

本文主要內容:
1 KEAP1缺乏的肺癌細胞對不同誘導劑誘導的鐵死亡有不同的影響
至少有3種不同類型的鐵死亡誘導劑,第一類誘導劑主要為erastin;第二類誘導劑為RSL3 和ML162,是通過抑制SLC7A11和GPX4RSL3引起鐵死亡;第三類誘導劑FIN56通過同時消耗GPX4蛋白和CoQ誘導鐵死亡(Fig. 1a)。在H1299細胞(KEAP1野生型肺癌細胞)中,KEAP1缺失顯著增加NRF2以及其的轉錄靶點SLC7A1138的水平(Fig 1b),并使H1299細胞對erastin引起的鐵死亡產生抗性(Fig 1c)。此外,在H1299細胞中敲除KEAP1后,GPX4水平顯著降低(Fig 1b)。盡管GPX4在KEAP1缺失的H1299細胞中表達下降,但KEAP1缺失顯著促進了對RSL3或ML162的抗鐵死亡的能力(Fig 1d)。相比之下,在H1299細胞中KEAP1卻并不影響鐵死亡對FIN56的敏感性(Fig 1e,f)。脂質過氧化是鐵死亡的一個標志。KEAP1的缺乏減弱了由erastin、RSL3或ML162引起的脂質過氧化,但對FIN56沒有影響。
在KEAP1缺失的H1299細胞中,GPX4水平的降低并不能解釋為什么這些細胞對RSL3具有耐藥性(抑制GPX4);另一方面,KEAP1缺失細胞中SLC7A11水平的增加可能是這些細胞對RSL3耐藥性增加的原因(Fig. 1b, d)。然而,作者發現在KEAP1敲除的H1299細胞中敲除SLC7A11對細胞中RSL3的鐵死亡敏感性沒有顯著影響(Fig. 1i, j)。使用ML162進行了類似的觀察(Fig 1k),并通過脂質過氧化測定進一步證實了這一觀察結果(Fig. 1l, m),然而,在H1299細胞中敲低GPX4誘導了大量脂質過氧化和鐵死亡,KEAP1敲低對應著GPX4缺失,沒有表現出明顯的脂質過氧化或者細胞死亡(Fig. 1n–q)。
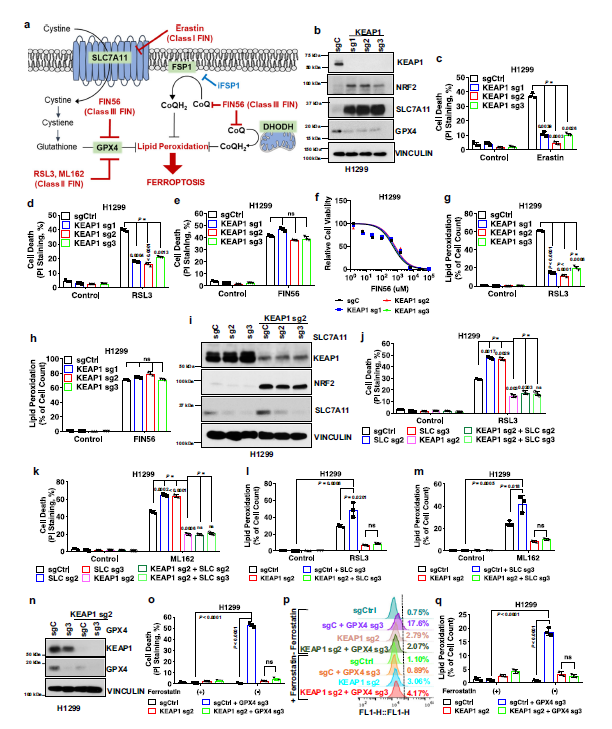
Fig1 KEAP1在肺癌細胞中以不依賴SLC7A11/ gpx4的方式調控鐵死亡
2. 在KEAP1缺失的肺癌細胞中,FSP1是2類和3類鐵死亡誘導劑差異效應的基礎
在KEAP1突變的肺腺癌中,發現12個過表達基因參與CoQ代謝過程,其中FSP1是上調最顯著的(Fig 2a)。KEAP1的缺失導致H1299和H23細胞中FSP1的表達增加(Fig 2b)。進一步表明,降低KEAP1敲除的H1299細胞中FSP1的表達(達到與對照細胞相似的水平)可使KEAP1缺失的細胞對RSL3或ML162重新敏感(Fig 2c–e)。在H1299和H23細胞中,KEAP1缺失誘導的對RSL3或ML162的抗性在iFSP1處理下基本消除(Fig. 2f, g)。在H1299細胞中,KEAP1的缺失降低了CoQ/CoQH2比值,而將KEAP1 KO細胞中的FSP1水平恢復到到對照組的水平,也可以恢復CoQ/CoQH2比值(Fig. 2h)。重要的是,在CoQ合成阻斷條件下,KEAP1缺陷誘導的肺癌細胞對RSL3或ML162的抗鐵死亡能力被大量消除(Fig. 2i–l)。總之,這些結果都表明KEAP1缺失可上調FSP1水平,并且在KEAP1缺陷的肺癌細胞中,CoQ-FSP1信號軸在介導鐵死亡對2類鐵死亡誘導劑的抵抗中發揮重要作用。
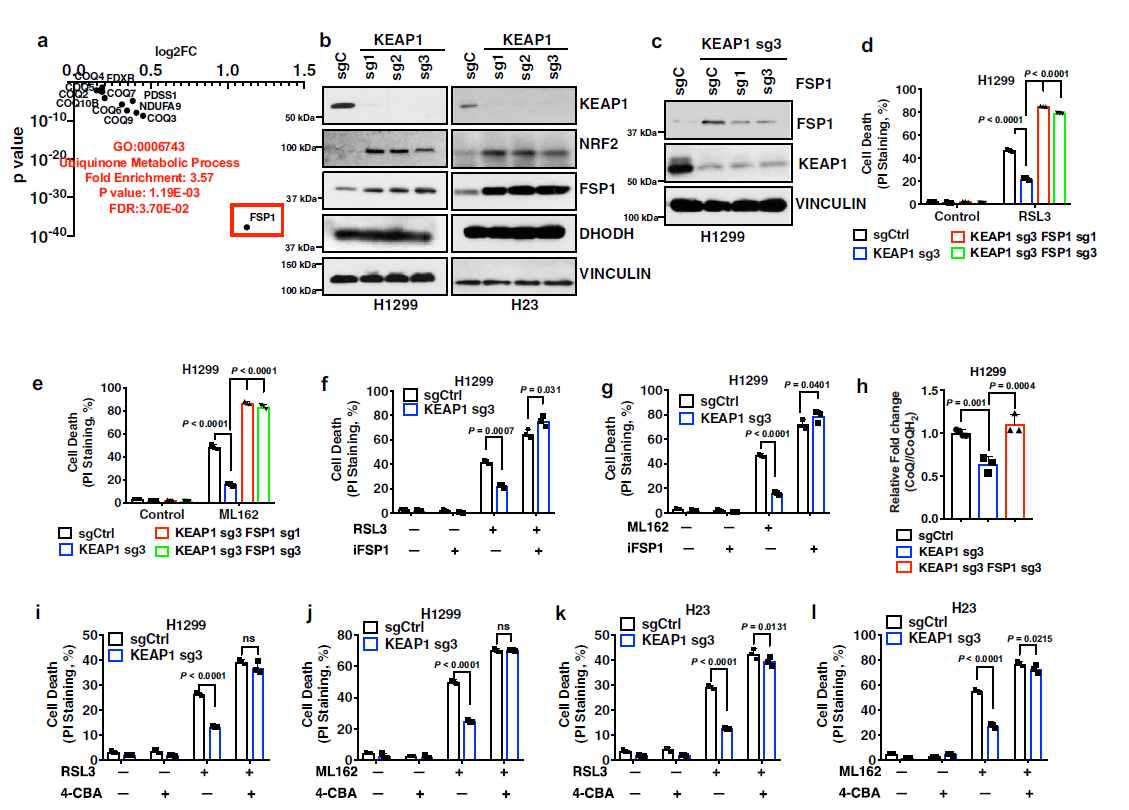
Fig2 KEAP1通過FSP1調控鐵死亡敏感性
3. KEAP1通過NRF2介導的轉錄調控FSP1
NRF2通常與抗氧化反應元素(AREs)結合在基因啟動子區域。進一步分析FSP1啟動子,發現在FSP1基因轉錄起始位點上游1kb范圍內發現了兩個AREs (Fig 3a)。Chip分析顯示,在A549細胞中,NRF2結合在兩個AREs上(Fig 3b, c)。同樣,在H1299細胞中將KEAP1刪除顯著增加了NRF2與這些AREs的結合(Fig 3d, e)。NRF2誘導劑叔丁基對苯二酚(TBHQ)的處理提高了NRF2和FSP1的水平(Fig. 3f)。相應的,FSP1啟動子-熒光素酶活性顯著增強,這一活性可以通過任意一個ARE的突變而部分降低,而兩個AREs的突變則幾乎完全消除(Fig. 3g, h)。同樣,KEAP1的缺失增加了FSP1啟動子熒光素酶的活性,而這種增加被ARE突變所消除(Fig. 3i)。
作者進一步研究了NRF2與KEAP1調控FSP1和鐵死亡的相關性。發現在KEAP1敲低的細胞中,消除NRF2可以抑制KEAP1缺陷誘導的FSP1啟動子熒光素酶活性和FSP1的表達 (Fig. 3i, j)。重要的是,在KEAP1/NRF2雙敲除的細胞中FSP1的重新表達(與KEAP1 KO細胞中相似的水平)恢復了這些細胞中的鐵死亡抗性(Fig. 3k, l)。綜上所述,這些數據表明FSP1是NRF2轉錄靶點,肺癌細胞中KEAP1缺失導致FSP1通過上調NRF2從而產生鐵死亡抗性。

Fig3 KEAP1通過nrf2介導的轉錄調控FSP1
4. 在KEAP1缺乏的肺癌中,FSP1對腫瘤生長至關重要
通過TCGA數據分析,作者研究了FSP1與人類癌癥的潛在相關性。發現FSP1在肺癌、腎透明細胞癌、腎乳頭狀細胞癌、腎癌、胃腺癌、肝癌終得表達量明顯高于正常人 (Fig. 4a)。此外,在LUAD、KIRC和卵巢癌中,FSP1的高表達與較短的患者生存期相關 (Fig. 4b)。FSP1缺失顯著抑制KEAP1 敲低的小樹中腫瘤的生長(Fig. 4c, d)。在這些腫瘤樣本中,脂質過氧化標記物4-HNE顯示KEAP1缺失降低了脂質過氧化標記物4-HNE的表達,在KEAP1缺失的腫瘤中,這被FSP1的缺失所恢復(Fig. 4e, f)。總之,在H1299異種移植模型中,FSP1對于KEAP1失活誘導的腫瘤生長是必需的(但可能是不夠的),此外,FSP1可能通過抑制脂質過氧化和鐵死亡促進KEAP1缺失的肺腫瘤生長。
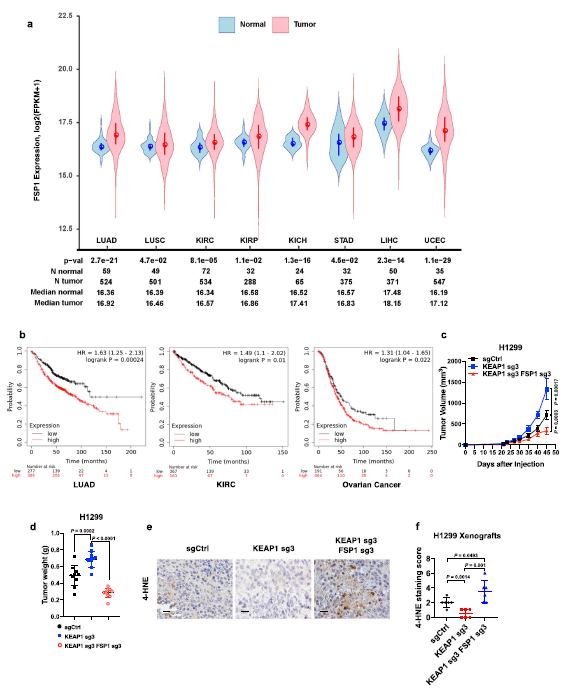
Fig4 FSP1在KEAP1缺陷癌中促進腫瘤發生
5. 抑制FSP1通過誘導鐵死亡使KEAP1缺陷的肺癌細胞對輻射敏感
作者發現,KEAP1的缺失顯著促進了H1299細胞的抗輻射能力,并進一步表明,NRF2的缺失使KEAP1敲除的細胞對放療重新敏感,而在KEAP1/NRF2雙敲除的細胞中重新表達FSP1可恢復這些細胞的抗輻射能力(Fig. 5a)。
KEAP1缺失顯著降低了放療誘導的脂質過氧化和鐵死亡標記基因PTGS2的表達;這種衰減在KEAP1敲除的細胞中通過NRF2的缺失而被消除,然后在KEAP1/NRF2雙敲除的細胞中通過FSP1的重新表達得以恢復(Fig. 5b, c)。作者進一步研究了FSP1失活是否使KEAP1缺失或突變的肺癌細胞對鐵死亡誘導劑敏感。作者發現,從基因上耗盡FSP1可使KEAP1敲除的 H1299細胞對化療敏感(Fig. 5d),并將化療誘導的KEAP1 敲低的細胞中脂質過氧化和PTGS2表達水平恢復到與對照細胞相似的水平(Fig. 5e, f)。同樣,iFSP1處理產生了強大的放射增敏效應(Fig. 5g),并促進了化療誘導的KEAP1缺乏的H1299細胞中脂質過氧化(Fig. 5h)。在KEAP1突變型A549細胞中,FSP1的缺失也促進了放射增敏和rt誘導的脂質過氧化(Fig. 5i–k)。通過在幾個KEAP1突變的肺癌細胞系中使用iFSP1獲得了類似的結果(Fig. 5l–o).
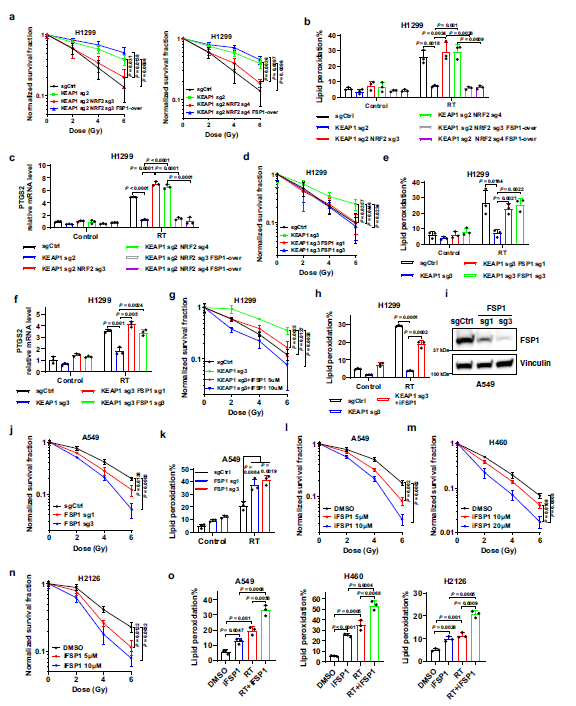
Fig5 FSP1抑制通過誘導鐵死亡使KEAP1缺陷的肺癌細胞對輻射敏感
6. 在KEAP1缺乏或突變的肺癌細胞或腫瘤中,抑制CoQ合成可以克服放射抗性
上述數據表明,FSP1是一個重要的治療靶點,iFSP1是治療KEAP1突變型癌癥的一種放射增敏劑。然而,iFSP1不能用于體內治療。由于FSP1與CoQ通過相同的途徑抑制亞鐵降解,作者進一步測試了抑制CoQ生物合成對KEAP1失活的肺癌細胞的放射敏感性是否有影響。結果發現,4-CBA治療逆轉了KEAP1缺失引起的放射耐藥(Fig. 6a),并在KEAP1 敲除 H1299細胞中恢復鐵死亡誘導劑誘導的脂質過氧化(Fig. 6b)。4-CBA處理對KEAP1突變體A549和H460細胞具有類似的放射增敏作用(Fig. 6c, d)。這些結果進一步證實了在A549和H460細胞中COQ2的基因缺失(Fig 6e–g)。重要的是,4-CBA處理沒有發揮放射增敏作用,也沒有促進化療誘導的人支氣管上皮細胞(HBECs)的脂質過氧化(Fig 6h, i);因此,與正常肺上皮細胞相比,4-CBA在KEAP1敲低的肺癌細胞中似乎具有更強的促亞鐵降解或放射增敏作用,提示4-CBA與化療聯合治療存在藥物治療窗。
在A549異種移植瘤模型中,4-CBA治療對腫瘤生長有中度抑制作用,4-CBA聯合RT治療對腫瘤生長有明顯抑制作用(Fig. 6j, k),與這些腫瘤樣本中4-HNE染色增加相關(Fig. 6l, m)。作者在具有KEAP1突變(TC494)的肺癌患者源性異種移植(PDXs)中進行了類似的觀察(Fig. 6n–q)。總的來說,研究表明可以通過使用4-CBA來克服KEAP1失活肺癌的放射耐藥。

Fig6抑制CoQ合成可逆轉KEAP1缺失或突變的肺癌細胞或腫瘤的放射耐藥
綜上所述,通過了解在KEAP1缺陷的肺癌細胞中不同類型的誘導劑引起的鐵死亡表型的機制,確定了CoQ-FSP1軸是一個關鍵的下游效應因子。在KEAP1缺乏的肺癌中,CoQ-FSP1軸作為KEAP1- nrf2通路的關鍵下游效應因子,介導ferroptosis和輻射抗性。作者進一步提出,利用CoQ-FSP1信號的藥理靶向可以克服KEAP1缺陷引起的放射耐藥,并治療KEAP1突變型肺癌。因此,作者的研究確定了一種治療這種致命疾病的潛在有效的治療策略。
參考文獻:
Koppula, P., et al., A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun, 2022. 13(1): p. 2206.