






適配器SH3BGRL通過促進乳腺癌中PIK3翻譯和ATG12穩定性來驅動自噬介導的化療耐藥
獲得性化療耐藥是乳腺癌復發的主要原因之一,但化療耐藥的潛在機制仍不清楚。目前,有研究指出一種小接頭蛋白SH3BGRL在乳腺癌患者中升高可能是通過自噬增強導致乳腺癌獲得性化療耐藥的關鍵驅動因素,而靶向SH3BGRL可能是一種潛在的治療策略。該研究于2021年12月發表在《Autophagy》,IF:16.016。
技術路線:

主要研究結果:
1. SH3BGRL上調與乳腺癌不良預后相關
與鄰近的正常乳腺癌相比,SH3BGRL mRNA在大多數乳腺癌中上調(圖1A,B),SH3BGRL mRNA水平升高的患者總體生存率和無復發生存率較差(圖1C-E)。此外,Kaplan Meier Plotter分析顯示,SH3BGRL蛋白含量高的乳腺癌患者的總生存期較差(圖1F),并且SH3BGRL蛋白在乳腺癌細胞株(圖1G)和乳腺癌組織中表達顯著上調(圖1H)。與正常乳腺細胞MCF-10A和配對的相鄰非癌組織相比,差異有統計學意義(p < 0.05)。免疫組化(IHC)發現與相鄰組織相比,SH3BGRL蛋白在乳腺癌組織中高表達(圖1I, J),驗證了SH3BGRL在乳腺癌中的表達上調。綜上所述,這些結果提示SH3BGRL上調可能是導致乳腺癌患者預后不良的原因之一。
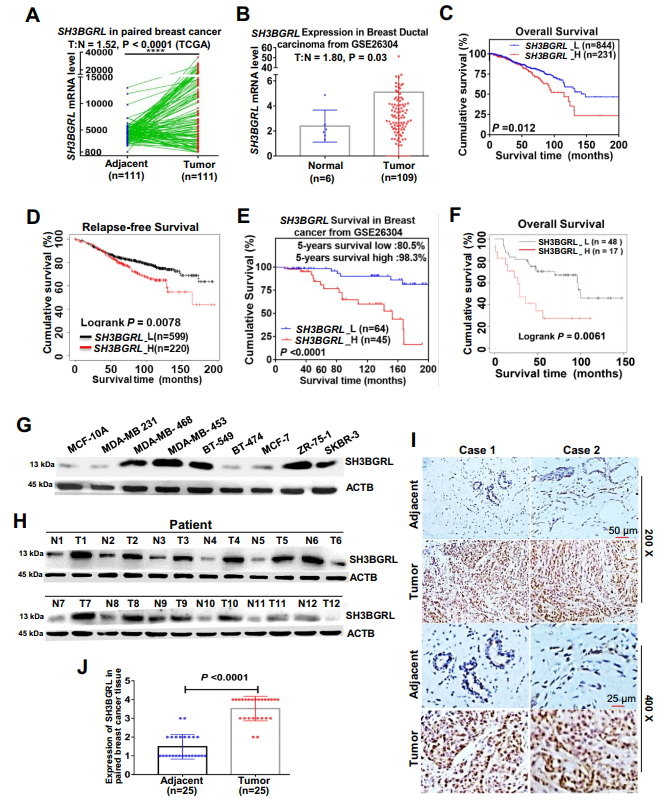
圖1 SH3BGRL上調與乳腺癌預后不良相關
2. SH3BGRL通過自噬保護作用對阿霉素細胞毒性產生耐藥性
為了了解SH3BGRL在乳腺癌進展和不良預后中的潛在生理作用,對來自各種公共數據集的乳腺癌患者隊列進行了基因集富集分析(GSEA)。GSEA分析表明SH3BGRL表達與阿霉素耐藥基因特征的富集呈正相關,但與阿霉素敏感基因特征呈負相關(圖2A)。通過對更多乳腺癌患者隊列(GSE45827和GSE15852)的分析,SH3BGRL表達也與自噬相關基因特征呈正相關(圖2B)。細胞毒性分析顯示,過表達SH3BGRL增加了細胞對阿霉素(Dox)的IC50,而敲低SH3BGRL則降低了IC50,而添加CQ抑制細胞自噬可有效中和這一效應(圖2C)。流式細胞術和集落形成分析證實,過表達SH3BGRL使細胞對Dox產生耐藥性,CQ或3-MA致敏細胞對Dox的細胞毒性阻斷細胞自噬(圖2D,E)。相比之下,抑制SH3BGRL使細胞凋亡對Dox處理敏感,降低了菌落形成能力,抑制自噬幾乎消除了這些差異(圖2D,E)。進一步評估了SH3BGRL過表達在皮下異種移植瘤模型中誘導的Dox耐藥性,發現即使使用Dox治療,SH3BGRL過表達也促進MCF-7細胞的腫瘤生長,而SH3BGRL沉默顯著增加MDA-MB-453細胞誘導的腫瘤對Dox的敏感性(圖2F-H)。因此,SH3BGRL可能具有自噬介導的乳腺癌化療耐藥性。
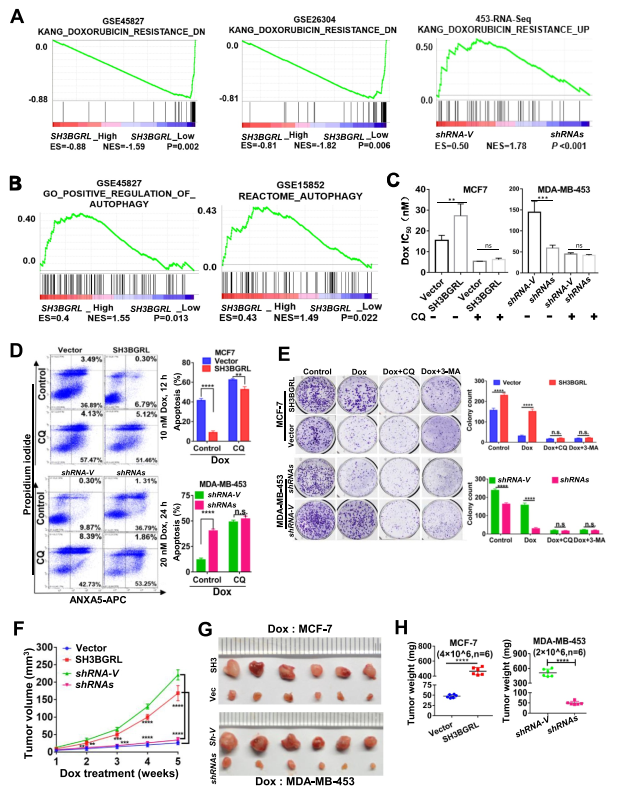
圖2 SH3BGRL通過自噬對阿霉素(Dox)誘導的凋亡產生抗性
3. SH3BGRL觸發乳腺癌細胞自噬
為了驗證SH3BGRL對大細胞自噬的驅動作用,檢測兩種典型的自噬標志物LC3B和SQSTM1,結果表明,即使在DOX誘導的應激下,過表達SH3BGRL也會降低SQSTM1水平。而SH3BGRL基因敲除增加了乳腺癌細胞的自噬進展(圖3A,B)以及相關的LC3B-I到LC3B-II的轉變(圖3C)。此外,當短時間抑制自噬體-溶酶體融合步驟與巴非霉素A1(Baf A1)以探測釋放的自噬通量時,可以觀察到在SH3BGRL過度表達的細胞中LC3B-II明顯富集,相反,在SH3BGRL敲除的細胞中LC3B-II褪色(圖3D,E),表明SH3BGRL誘導自噬通量。免疫熒光染色顯示,在MCF-7細胞中,SH3BGRL過表達后LC3B聚集點明顯增加,而在MDA-MB -453細胞中,在無血清或無氨基酸的EBSS饑餓條件下,SH3BGRL敲除后LC3B聚集點明顯減少(圖3F-H),也證實了溶酶體SQSTM1的顯著降解(圖4A),并且沒有明確的MTOR激活與SH3BGRL誘導的自噬有關(圖4B)。此外,IHC染色和統計分析表明,與配對的相鄰正常組織相比,SH3BGRL高表達患者的總LC3B蛋白水平明顯降低(圖4C,D)。同時,患者的SH3BGRL蛋白水平與LC3B蛋白水平呈負相關(圖4E)。總之,這些結果表明SH3BGRL升高促進乳腺癌細胞的自噬進展。
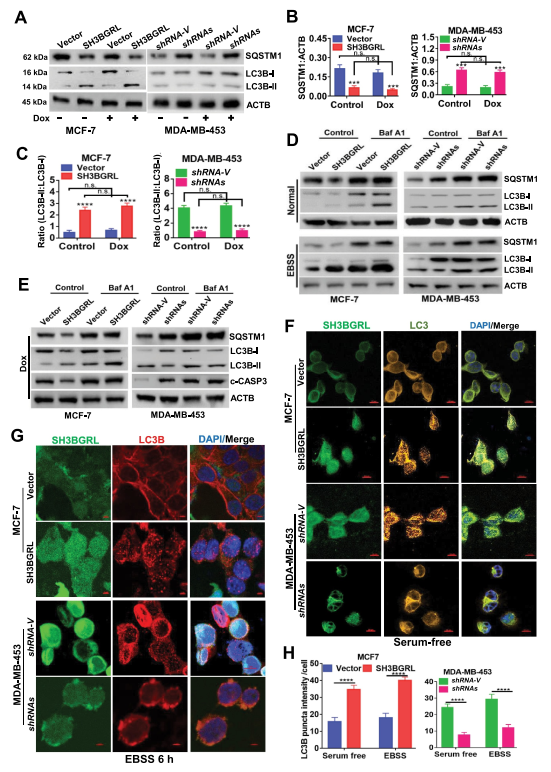
圖3 SH3BGRL促進乳腺癌細胞的自噬通量
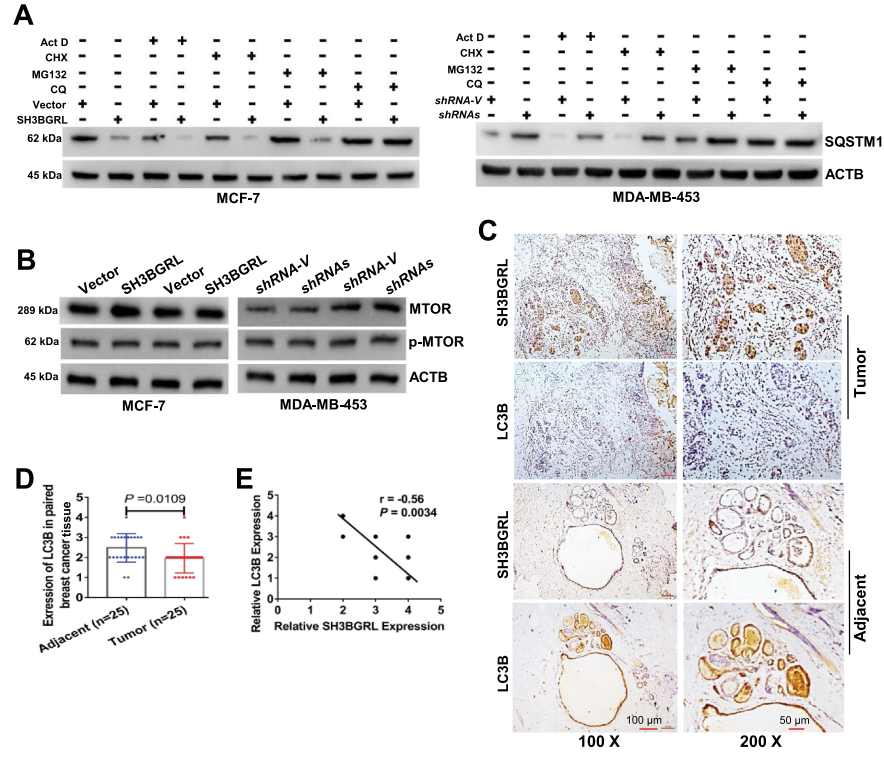
圖4 SH3BGRL可使乳腺癌發生自噬
4. SH3BGRL通過與核糖體相互作用,通過PIK3C3翻譯增強來驅動自噬
質譜分析法(MS)鑒定顯示SH3BGRL與大量的翻譯機制蛋白相互作用(圖5A)。分析序列中自噬相關基因的翻譯效率,發現自噬啟動的PIK3C3 mRNA向較輕的部分平移約4次,表明其翻譯衰減(圖5B)。免疫印跡結果顯示,PIK3C3在過表達SH3BGRL的細胞中明顯升高,而在敲除SH3BGRL的細胞中明顯降低(圖5C)。當翻譯抑制劑環己酰亞胺(CHX)阻斷核糖體翻譯后,過表達SH3BGRL的細胞PIK3C3蛋白水平明顯恢復到親本細胞水平(圖5D)。進一步在細胞中進行了多核糖體分析,并觀察到PIK3C3 mRNA在SH3BGRL高細胞中轉移到較重的多核糖體部分(圖5E),而在SH3BGRL敲除細胞中,PIK3C3 mRNA又轉移到較輕的部分(圖5F)。ATG12(圖5G)和其他自噬基因的蛋白質變化(圖5H)沒有部分移位或轉錄表達變化。綜上所述, SH3BGRL至少可以通過與核糖體的相互作用促進PIK3C3的翻譯,從而促進乳腺癌細胞自噬。
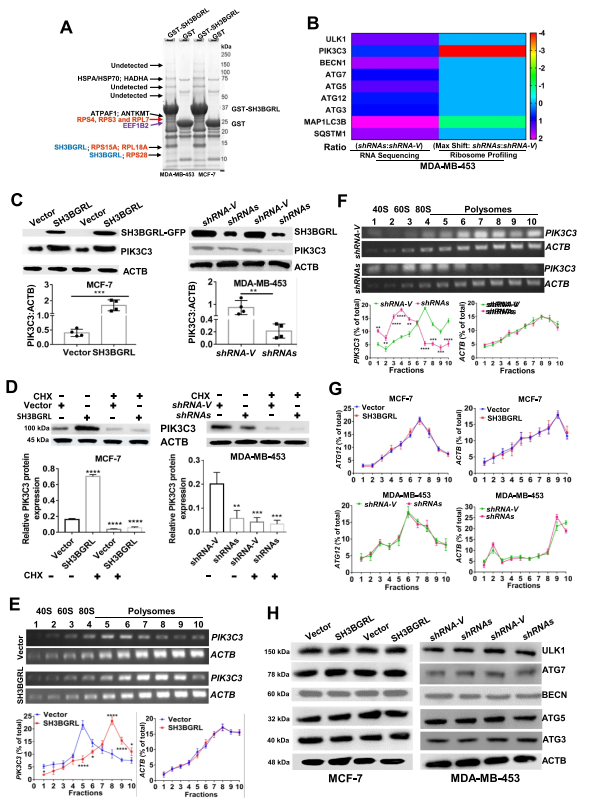
圖5 SH3BGRL通過核糖體相互作用,通過PIK3C3翻譯,驅動自噬
5. SH3BGRL也通過維持ATG12的穩定性來促進自噬
SH3BGRL在蛋白質水平上有效地上調乳腺癌細胞中的ATG12 (圖6A),但其mRNA水平和所述的翻譯事件不起作用。CHX追蹤實驗表明,SH3BGRL充分降低了MCF-7細胞中ATG12的降解率,而SH3BGRL沉默則加速了MDA-MB-453細胞中ATG12的降解率(圖6B)。免疫共沉淀(co-IP)顯示SH3BGRL與ATG12和BECN相互作用,但在所有細胞中沒有其他自噬相關蛋白(圖6C)。為了進一步研究較少SH3BGRL突變體的ATG12可能的蛋白酶體降解,用蛋白酶體抑制劑MG132處理細胞,發現SH3BGRL中的α3螺旋或β3片對SH3BGRL與ATG12的結合仍然至關重要(圖6D)。免疫印跡結果顯示,阻斷蛋白酶體過程、在親本MCF-7細胞或MDA-MB-453 SH3BGRL敲除細胞中ATG12的表達水平與高SH3BGRL的對應細胞相似(圖6E),表明原生體抑制對SH3BGRL的模擬效應。泛素化分析直接表明SH3BGRL過表達細胞中泛素結合的ATG12減少,而SH3BGRL沉默細胞中泛素結合的ATG12增加(圖6F)。因此,這些結果表明SH3BGRL可以通過與ATG12相互作用阻止其降解或通過結合并削弱乳腺癌細胞中的ATG12 E3連接酶功能來穩定ATG12,從而平行增強自噬。
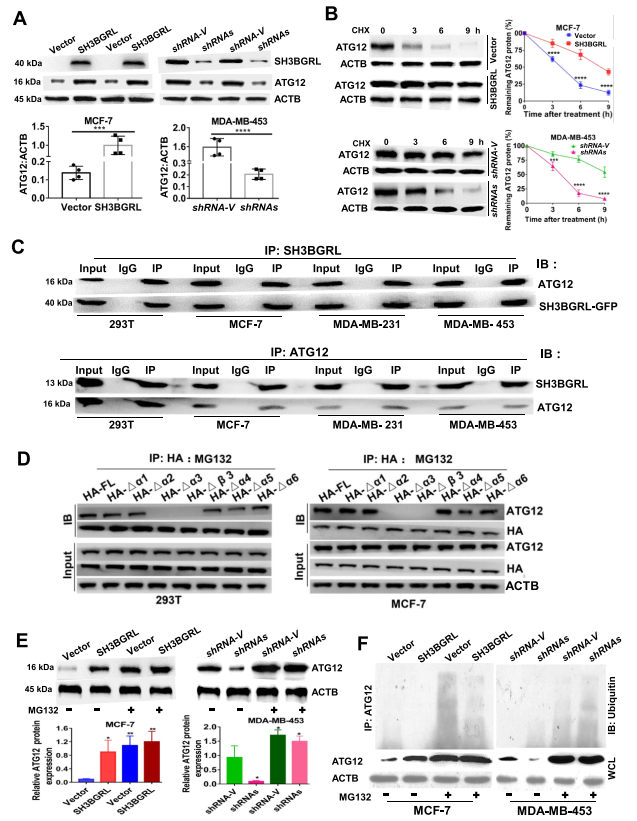
圖6 SH3BGRL也通過維持ATG12的穩定性來增強自噬
6. PIK3C3和ATG12有助于SH3BGRL介導的阿霉素耐藥
為了驗證SH3BGRL-PIK3C3/ATG12軸所表征的自噬介導的Dox抗性,我們分別在SH3BGRL過表達細胞中敲除PIK3C3或ATG12(圖7A),或在SH3BGRL敲除細胞中強制表達它們(圖7B)。流式細胞術分析顯示,在SH3BGRL-overexpressing MCF-7的細胞中,內源性PIK3C3或ATG12的額外沉默可顯著促進細胞凋亡(圖7C),自噬減少(圖7D),同時在Dox作用下,PARP和CASP3切割導致細胞凋亡升高(圖7E)。同時,這種PIK3C3或ATG12沉默導致細胞活力降低,Dox處理下菌落形成減少(圖7F-J)。相反,在SH3BGRL敲低MDA-MB-453細胞中強制表達PIK3C3或ATG12可抑制細胞凋亡,增加自噬水平和Dox耐藥(圖7A-J),證實自噬在SH3BGRL誘導的化療耐藥中對PIK3C3或/和ATG12上調的重要作用。
通過皮下移植瘤模型進一步評價了SH3BGRL介導的自噬對乳腺癌Dox耐藥的生理作用。乳腺癌細胞中,敲低SH3BGRL使腫瘤對Dox治療顯著敏感,導致腫瘤負荷降低,而過表達PIK3C3或ATG12則使腫瘤消退消失(圖8A, B)。Dox與自噬抑制劑CQ聯合使用可以有效地使MDA-MB-453細胞中SH3BGRL高腫瘤的消退(圖8A,B)。去除SH3BGRL能充分抑制Dox作用下的細胞自噬和腫瘤細胞凋亡(圖8C,D)。而在SH3BGRRL沉默的腫瘤中,過表達PIK3C3或ATG12可促進自噬,抑制腫瘤細胞凋亡,Dox與CQ聯合使用可促進更多的腫瘤細胞凋亡(圖8C,D)。此外,SH3BGRL對其他化療耐藥基因沒有明顯影響,表明SH3BGRL衍生的自噬是化療的主要原因(圖8E,F)。總之,這些結果表明SH3BGRL-PIK3C3-ATG12自噬軸賦予乳腺癌有效的Dox或其他化療耐藥。

圖7 PIK3C3和ATG12參與了SH3BGRL介導的阿霉素耐藥

圖8 PIK3C3和ATG12有助于致瘤性和Dox耐藥
7. 乳腺癌患者SH3BGRL-PIK3C3/ATG12典型自噬的激活
接下來研究SH3BGRL-PIK3C3/ATG12自噬在乳腺癌中的臨床意義。對乳腺癌標本進行SH3BGRL、PIK3C3、ATG12和LC3B的免疫組化染色。結果顯示,與配對的相鄰正常組織相比,SH3BGRL、ATG12和PIK3C3在乳腺癌中協同升高,而LC3B下調(圖9A-C)。統計分析顯示,SH3BGRL與PIK3C3、ATG12表達呈正相關,與LC3B表達呈負相關(圖9D)。免疫印跡進一步驗證, SH3BGRL表達與PIK3C3和ATG12呈正相關,與SQSTM1和LC3B呈負相關(圖9E-G)。PIK3C3和ATG12 mRNA水平的小于1.2倍的邊緣變化也通過其他乳腺癌隊列(GSE45827、GSE26304和TCGA數據集)的分析來驗證(圖9H)。Kaplan Meier Plotter表明,PIK3C3或ATG12水平高的患者乳腺癌患者的5年總生存期、無復發生存期和無轉移生存期明顯較差(圖9I)。以上所有結果通過乳腺癌轉移復發和較差臨床結果的轉錄后調節,驗證了SH3BGRL-PIK3C3-ATG12自噬軸與乳腺癌Dox化療耐藥的相關性(圖9J)。
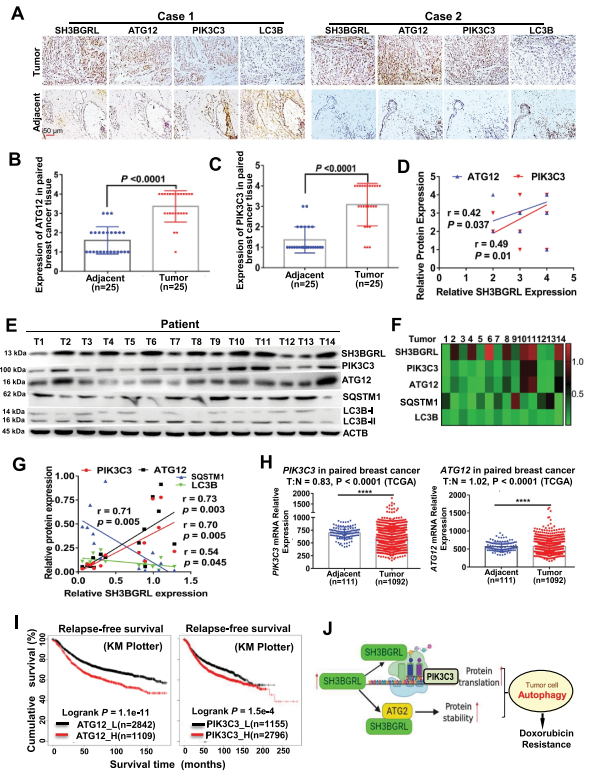
圖9 乳腺癌患者SH3BGRL-PIK3C3/ATG12自噬軸的激活
主要結論:
該研究結果至少揭示了SH3BGRL是一種新型耐藥基因,通過上調PIK3C3和ATG12,促進乳腺癌自噬介導的Dox耐藥。因此,靶向SH3BGRL或其下游關鍵效應因子將是克服乳腺癌化療耐藥性的可能策略。