






巨噬細胞已經(jīng)過時了嗎?非也,看過來學習新思路
腫瘤相關(guān)巨噬細胞(TAMs)支持腫瘤進展在響應(yīng)腫瘤和基質(zhì)細胞構(gòu)成的微環(huán)境中扮演著促腫瘤表型作用。但是腫瘤細胞指示TAM行為的潛在機制仍然未知。該文發(fā)現(xiàn)腫瘤-細胞-來源的葡萄糖神經(jīng)酰胺刺激的非傳統(tǒng)性內(nèi)質(zhì)網(wǎng)壓力響應(yīng)通過誘導巨噬細胞內(nèi)質(zhì)網(wǎng)膜的脂質(zhì)組分重組和飽和,這誘導了IRE1-介導的XBP1剪切和STAT3激活。這種腫瘤-細胞-產(chǎn)生的脂質(zhì)通過內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng)同時協(xié)調(diào)巨噬細胞極化和腫瘤存活的作用,可能是維持宿主抗腫瘤免疫的治療靶點。本文于2021年10月發(fā)表在《Nature Immunology》IF: 25.606雜志上。
技術(shù)路線:

主要實驗結(jié)果:
1、TAMs表現(xiàn)出高脂質(zhì)含量和ER應(yīng)激反應(yīng)
為了了解TAM的脂質(zhì)代謝,作者在YUMM1.7黑素瘤小鼠移植模型和基因工程小鼠黑素瘤模型(簡稱Braf/Pten小鼠)中,我們通過bodypy和FilipinIII染色和bodypy C12(一種熒光脂類類似物)攝取來評估腫瘤和脾臟中巨噬細胞的脂質(zhì)含量和攝取。在這兩種模型中,與脾臟巨噬細胞相比,TAMs增加了中性脂質(zhì)(BODIPY染色)和膽固醇(Filipin染色)含量和攝取(圖1a,b)。此外,脂滴只出現(xiàn)在TAM的細胞質(zhì)中,而不出現(xiàn)在脾臟巨噬細胞中(圖1c)。致瘤功能的標記酶ARG1陽性的巨噬細胞的脂質(zhì)含量高于ARG1陰性的TAMs(圖1d),提示TME引起的異常脂質(zhì)積累可能會扭曲TAMs的致瘤特性。除了脂滴的形成外,我們還發(fā)現(xiàn)TAMs的內(nèi)質(zhì)網(wǎng)更加擴張和腫脹,這是內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng)的形態(tài)學標志(圖1e)。以上表明TAMs具有更高的脂質(zhì)含量和ER應(yīng)激反應(yīng)。
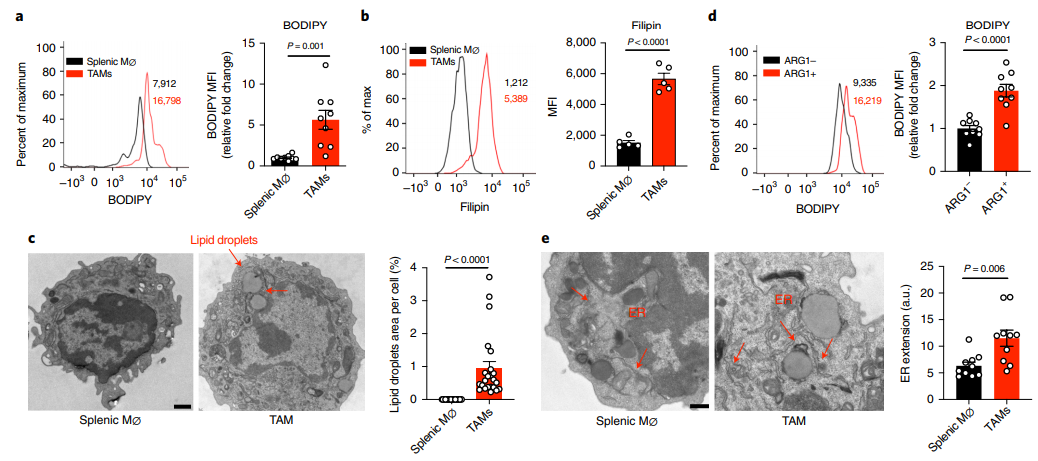
圖1 TMA促進致瘤性TAMs的脂質(zhì)積累
進一步支持了上述結(jié)論,研究發(fā)現(xiàn)TAMs增加了典型內(nèi)質(zhì)網(wǎng)逆境應(yīng)答基因的mRNA表達,包括Ern1, Bip和剪接Xbp1 (sXBP1),無論是在移植還是誘導的黑色素瘤模型中(圖2a),并且,與脾臟巨噬細胞相比,TAMs中sXBP1蛋白的表達率更高(圖2b),表明TAMs可能參與內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng)。由于最近的研究表明,脂質(zhì)代謝失調(diào)和內(nèi)質(zhì)網(wǎng)應(yīng)激之間的相互干擾在調(diào)節(jié)代謝組織的細胞行為方面發(fā)揮了重要作用,作者接下來檢測了脂質(zhì)含量sXBP1和ARG1在TAMs中的表達。結(jié)果顯示sXBP1+ TAMs 具有更高的脂質(zhì)含量(圖2c)和更高組分的致瘤性ARG1+ TAMs(圖2d)。通過計算幾種人類癌癥,包括大腸癌、無慢性阻塞性肺疾病的肺腺癌(LUAD-No COPD)和非小細胞肺癌(NSCLC)和一組小鼠肉瘤的單細胞RNA測序結(jié)果中單個TAM的內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng),作者觀察到,與抗腫瘤TAMs相比,不同癌癥中的促腫瘤TAMs都表現(xiàn)出更高的內(nèi)質(zhì)層應(yīng)激得分(圖2e)。綜上所述,內(nèi)質(zhì)網(wǎng)應(yīng)激和脂質(zhì)代謝失調(diào)可能相互協(xié)調(diào),從而誘導TAMs的致瘤性特征。
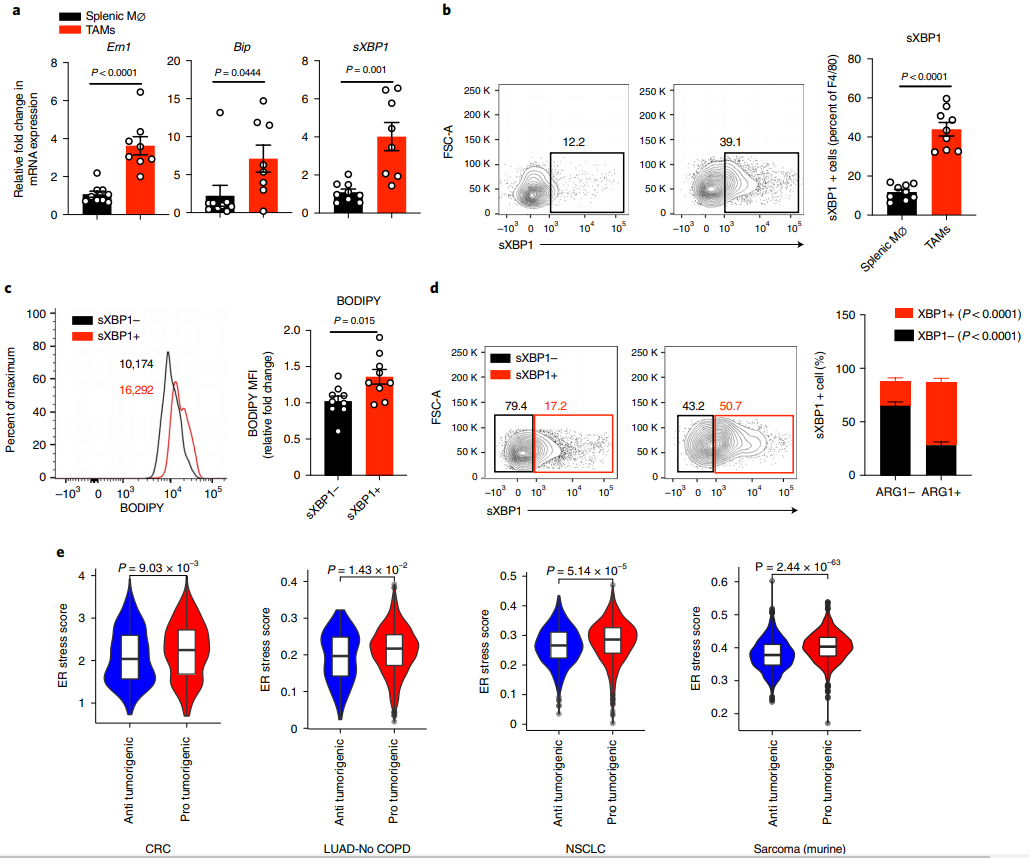
圖2 TME激活致瘤性TAMs的IRE1/sXBP1通路
2、IRE1-XBP1增強巨噬細胞腫瘤前極化
為了探究是否腫瘤-細胞-來源因子可以刺激增加巨噬細胞的脂質(zhì)含量和內(nèi)質(zhì)網(wǎng)應(yīng)激,作用用黑色素瘤YUMM1.7腫瘤-細胞-來源的條件培養(yǎng)基(CM)處理骨髓來源巨噬細胞(BMDM)。結(jié)果顯示,與對照相比,CM顯著促進BMDM的脂質(zhì)含量和攝取(圖3a),相應(yīng)的,促腫瘤標志物基因的表達也顯著增加,如Arg1和Mrc1(圖3b)。與na?ve巨噬細胞相比,CM處理的BMDM具有更高的抑制CD8+ T細胞增殖的活性
此外,CM處理的BMDM表現(xiàn)出sXBP1的產(chǎn)生,但是PERK的激活和下游靶基因ATF4表達少量增加(圖3c)。鑒于內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng)最近被揭示通過調(diào)節(jié)CD8+ T細胞、樹突狀細胞和MDSCs的功能來抑制抗腫瘤免疫,作者推測CM介導的IRE1-XBP1信號的激活可能支持了巨噬細胞獲得致瘤表型。為了驗證這一假設(shè),用STF081030處理CM刺激的BMDM,STF081030是一種抑制IRE1的核糖核酸內(nèi)切酶活性的抑制劑,可以防止sXBP1的產(chǎn)生,結(jié)果發(fā)現(xiàn)STF081030有效地抑制了由CM引起的sXBP1和促腫瘤標志物基因的表達(圖3d)。此外,STF081030處理有效改善了CM對BMDM的抑制能力(圖3e)。
為了證實IRE1在誘導前致瘤極化中的作用,將LysM-Cre Cas9小鼠產(chǎn)生的BMDM分別用含有scramble guide RNAs (gRNAs)或IRE1靶向gRNAs的慢病毒轉(zhuǎn)導,以產(chǎn)生對照BMDM或IRE1缺陷BMDM。與對照組BMDM相比,IRE1缺陷BMDM中IRE1表達較低,研究發(fā)現(xiàn)CM誘導的致瘤前極化在IRE1缺陷BMDM中被抑制(圖3f),這表明CM誘導的IRE1活性增強了巨噬細胞中致瘤前極化。傳統(tǒng)的內(nèi)質(zhì)網(wǎng)應(yīng)力誘導劑,包括tunicamycin和thapsigargin在內(nèi),均能誘導內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng),但不能使BMDM分化為致瘤表型(圖3g)。以上表明,與傳統(tǒng)內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng)相比,sXBP1控制的特殊信號級聯(lián)反應(yīng)是加強巨噬細胞對腫瘤細胞衍生因子反應(yīng)的免疫抑制活性所必需的。

圖3 腫瘤細胞通過IRE1在BMDM中驅(qū)動致瘤前極化
3、XBP1重塑TAM表型并支持腫瘤進展
為了進一步闡明XBP1對巨噬細胞致瘤前極化的貢獻,將LysM-Cre Cas9小鼠產(chǎn)生的BMDM分別用含有雜合GRNAS或XBP1靶向GRNAS的慢病毒轉(zhuǎn)導,以生成對照BMDM或XBP1缺陷BMDM。與對照組相比,XBP1缺陷BMDM中XBP1總表達和剪接表達均降低。結(jié)果發(fā)現(xiàn),與對照組BMDM相比,XBP1缺陷BMDM中CM誘導的致瘤標志物基因表達減少(圖4a)。此外,與XBP1野生型小鼠相比,CM對小鼠的免疫抑制能力在XBP1缺失后顯著消失,表現(xiàn)為免疫細胞數(shù)增多(圖4b),并且XBP1缺失小鼠的腫瘤體積和重量也都顯著下降(圖4c, d),提示XBP1可使巨噬細胞向致瘤前表型傾斜。作者通過檢測WT和XBP1cko小鼠的TAMs,發(fā)現(xiàn)XBP1缺失導致了mTAMs和iTAMs豐度的變化,以及iTAMs與mTAMs的比例降低(圖4e),表明敲除XBP1可以改善TAMs的致瘤性特征。總之,XBP1的表達促進TAMs向致瘤表型激活。
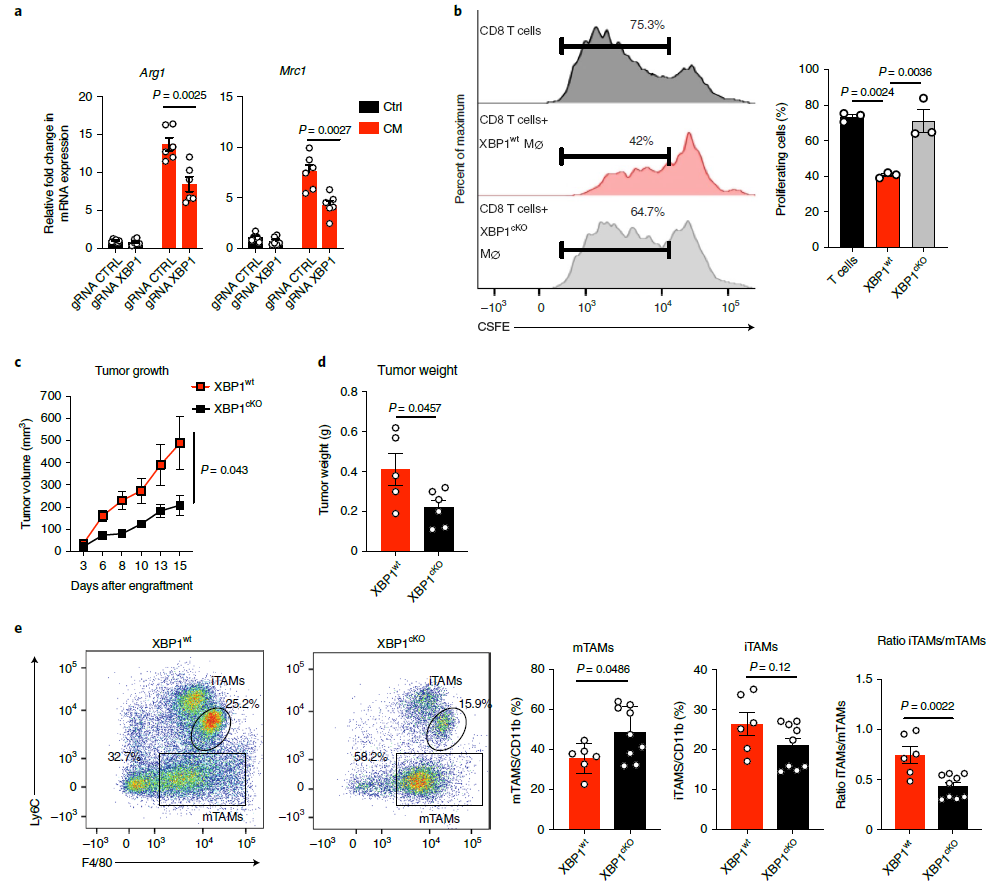
圖4 TAMs中XBP1缺失抑制腫瘤生長
4、STAT3信號優(yōu)化巨噬細胞腫瘤前極化
接下來,探究sXBP1單獨表達是否足以促進M2表型。結(jié)果顯示,STF083010處理顯著抑制了對照BMDM細胞中CM誘導的致瘤性標志物基因的表達,但對過表達sXBP1的BMDM沒有影響(圖5a)。支持了此前的結(jié)論,即在CM刺激的巨噬細胞中,需要IRE1介導的sXBP1的產(chǎn)生來增強原致瘤活性。
研究表明,STAT3的激活通過觸發(fā)TAMs中的組織蛋白酶表達來促進腫瘤進展。作者發(fā)現(xiàn),CM處理顯著增加了STAT3的磷酸化(圖5b)。推測,STAT3可能也是增強腫瘤-細胞CM的致瘤極性所需要的。研究顯示STAT3的基因缺失或STAT3抑制劑Stattic的處理會降低Mrc1基因的表達,但不會影響Arg1的表達(圖5c),這表明STAT3的激活可能在一定程度上參與了CM處理BMDM的抑制活性。接下來,檢測了腫瘤-細胞是否可以通過IL-6和IL-10的產(chǎn)生激活BMDM中的STAT3。結(jié)果發(fā)現(xiàn)抗體IL - 6和IL - 10無法抑制CM-Induced STAT3磷酸化(圖5d)和致瘤性標記基因的表達(圖5e),而該劑量的抗體能夠廢除導IL - 6和IL – 10誘導的STAT3磷酸化(圖5d)。些結(jié)果表明,腫瘤-細胞-來源因子激活STAT3,以IL-6/IL-10不依賴的方式促進巨噬細胞的致瘤前極化。
IRE1已被證明通過與STAT3形成蛋白復合物來支持肝細胞中STAT3的激活。通過使用鄰近連接試驗,發(fā)現(xiàn)CM促進了BMDM中的IRE1-STAT3相互作用(圖5f)。然后,檢測STAT3磷酸化是否需要IRE1,發(fā)現(xiàn)與對照組BMDM相比,腫瘤細胞CM誘導的STAT3磷酸化在IRE1缺陷的BMDM中被抑制(圖5g)。總之,以上結(jié)果表明CM觸發(fā)的IRE1信號通過同時刺激STAT3磷酸化和sXBP1的產(chǎn)生,導致巨噬細胞的致瘤極化。
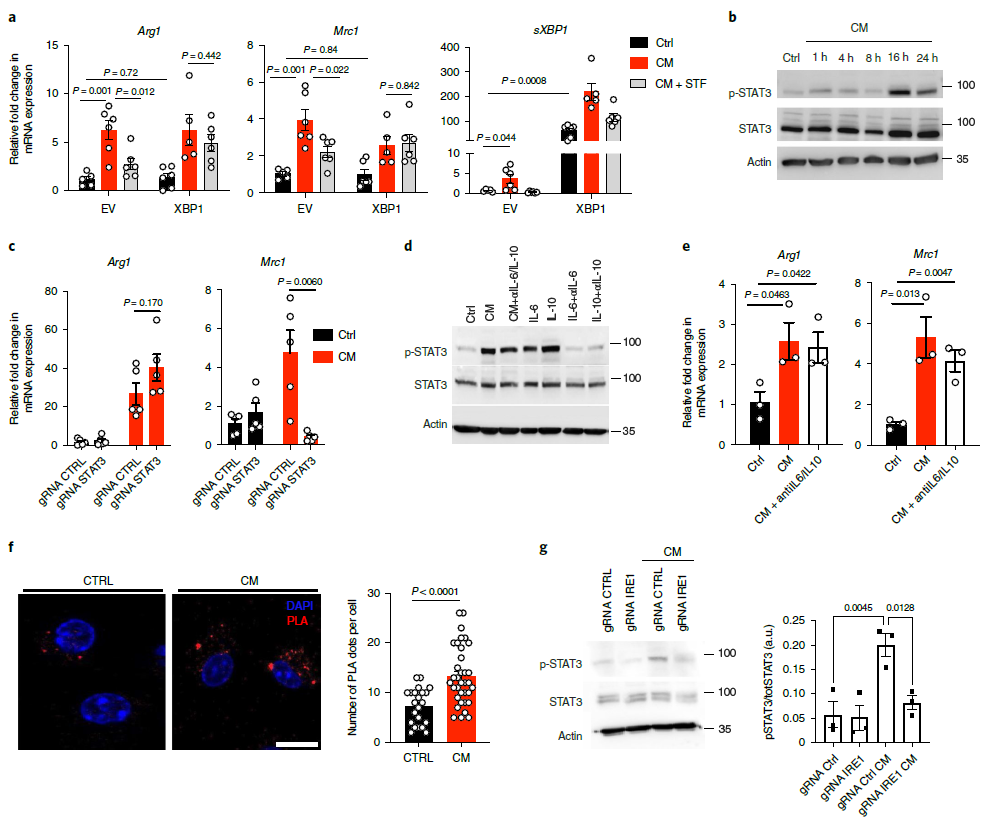
圖5激活I(lǐng)RE1-STAT3信號支持CM誘導極化
5、Mincle感知葡萄糖神經(jīng)酰胺使巨噬細胞活化
由于脂質(zhì)代謝異常被認為是誘發(fā)內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng)的原因,作者推測,腫瘤細胞產(chǎn)生的脂質(zhì)可能是誘導巨噬細胞致瘤前特征的原因。為了驗證這一點,使用脂質(zhì)去除劑從YUMM1.7黑色素瘤細胞CM中去除脂質(zhì),包括膽固醇和脂肪酸。結(jié)果發(fā)現(xiàn)CM脂質(zhì)去除后,CM引發(fā)事件的能力也都消失了,包括sXBP1產(chǎn)生、STAT3磷酸化和致瘤標志物基因的表達(圖6a-c)。檢測CM和脂質(zhì)去除CM的轉(zhuǎn)錄組,發(fā)現(xiàn)巨噬細胞中大多數(shù)負責致瘤活性的基因以脂依賴的方式在腫瘤細胞CM中上調(diào)(圖6d)。腫瘤細胞產(chǎn)生的脂質(zhì)可能控制內(nèi)質(zhì)網(wǎng)應(yīng)激參與的巨噬細胞致瘤性極化。
通過檢查轉(zhuǎn)錄組分析,作者重點關(guān)注參與脂質(zhì)基因識別和綁定的,并在CM組顯著上調(diào)的基因,發(fā)現(xiàn)基因——巨噬細胞誘導Ca2+依賴外源凝集素受體(Mincle),也稱為C型凝集素域家族4成員E (Clec4e),顯著響應(yīng)CM處理(圖6e)。進一步研究發(fā)現(xiàn),CM對BMDM的免疫抑制,上調(diào)致瘤性標志基因表達,和STAT3磷酸化的作用,都能被anti-Mincle抗體所反轉(zhuǎn)(圖6f-h)。總之,這些結(jié)果表明,Mincle介導的脂質(zhì)識別是CM誘導巨噬細胞致瘤前極化所需要的。
由于β-葡萄糖神經(jīng)酰胺和膽固醇硫酸鹽是Mincle已知的內(nèi)源性配體,所以作者研究了YUMM1.7細胞中β-葡萄糖神經(jīng)酰胺的產(chǎn)生是否是BMDM中驅(qū)動CM誘導的致瘤前極化所需要的。構(gòu)建針對UDP -葡萄糖神經(jīng)酰胺葡萄糖基轉(zhuǎn)移酶(UGCG)的shRNA,UGCG是β-葡萄糖神經(jīng)酰胺生成的代謝酶,以減少YUMM1.7黑色素瘤細胞中UGCG表達和β-葡萄糖神經(jīng)酰胺生成(圖6i)。結(jié)果顯示,和對照相比,UGCG缺失的CM對巨噬細胞致瘤性誘導的能力顯著下降,表現(xiàn)為UGCG缺失組的致瘤性標志物基因表達下降,STAT3磷酸化下降,腫瘤的體積和重量下降,iTAM/mTAM比值下降(圖6j-n)。總之,這些結(jié)果表明,腫瘤細胞產(chǎn)生的β-葡萄糖神經(jīng)酰胺以Mincle依賴的方式觸發(fā)內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng),釋放TAMs的致瘤活性。
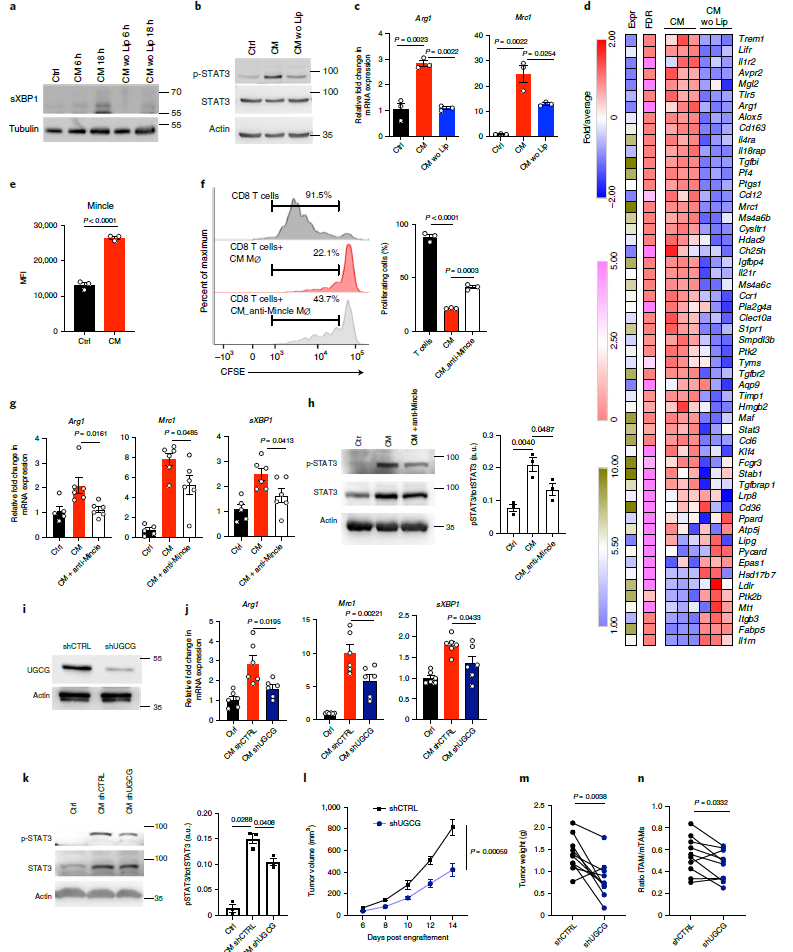
圖6 Mincle依賴的葡萄糖神經(jīng)酰胺感知途徑使巨噬細胞活化
6、內(nèi)質(zhì)網(wǎng)膜脂質(zhì)成分紊亂激活sXBP1
鑒于文獻表明Mincle可以抑制膽固醇外排,以及TAM可以積累更多的膽固醇,所以作者推測CM可能是促進細胞內(nèi)膽固醇積累通過刺激膽固醇合成。Fillipin染色結(jié)果表明CM可以促進細胞內(nèi)膽固醇含量,但是他汀處理阻斷膽固醇合成以及anti-Mincle抗體處理都可以有效的阻礙CM誘導的膽固醇積累(圖7a)。此外,他汀類藥物阻斷膽固醇合成也阻止了CM誘導的sXBP1產(chǎn)生和致瘤性標志物基因在BMDM中的表達,但是他汀類藥物和anti-Mincle抗體協(xié)同作用不會進一步放大該效應(yīng)(圖7b)。IRE1含有一個跨膜結(jié)構(gòu)域,可以感知脂質(zhì)失衡并誘導其二聚和激活,增加膽固醇積累可能會使內(nèi)質(zhì)網(wǎng)膜的脂質(zhì)成分重組為較低的磷脂酰膽堿與磷脂酰乙醇胺比值(PC/PE比值)和較低的多不飽和脂肪酸(PUFA),它會降低內(nèi)質(zhì)網(wǎng)的膜流動性,這就是膽固醇感應(yīng)機制紊亂的結(jié)果。因此,作者推測腫瘤-細胞CM通過重組內(nèi)質(zhì)網(wǎng)膜上的脂質(zhì)組分來觸發(fā)IRE1/XBP1的激活。內(nèi)質(zhì)網(wǎng)膜脂質(zhì)譜顯示,與對照組相比,CM處理的BMDM的PC/PE比值顯著降低,并觀察到PUFA的比值顯著下降(圖7d, e)。隨后,作者通過過表達LPCAT3(一種PC合成酶)試圖挽救內(nèi)質(zhì)網(wǎng)膜脂質(zhì)組分。結(jié)果發(fā)現(xiàn),過表達LPCAT3顯著增加了CM刺激的內(nèi)質(zhì)網(wǎng)膜的PC/PE比值和不飽和PC的豐度(圖7f-h),還觀察到LPCAT3過表達減少了CM處理的BMDM內(nèi)質(zhì)網(wǎng)膜的延伸(圖7i),以及阻斷CM誘導的sXBP1產(chǎn)生和致瘤性基因的表達(圖7j-l)。綜上所述,這些數(shù)據(jù)表明,腫瘤誘導的內(nèi)質(zhì)網(wǎng)膜脂質(zhì)成分重組對啟動內(nèi)質(zhì)網(wǎng)應(yīng)激介導的致瘤極化至關(guān)重要。
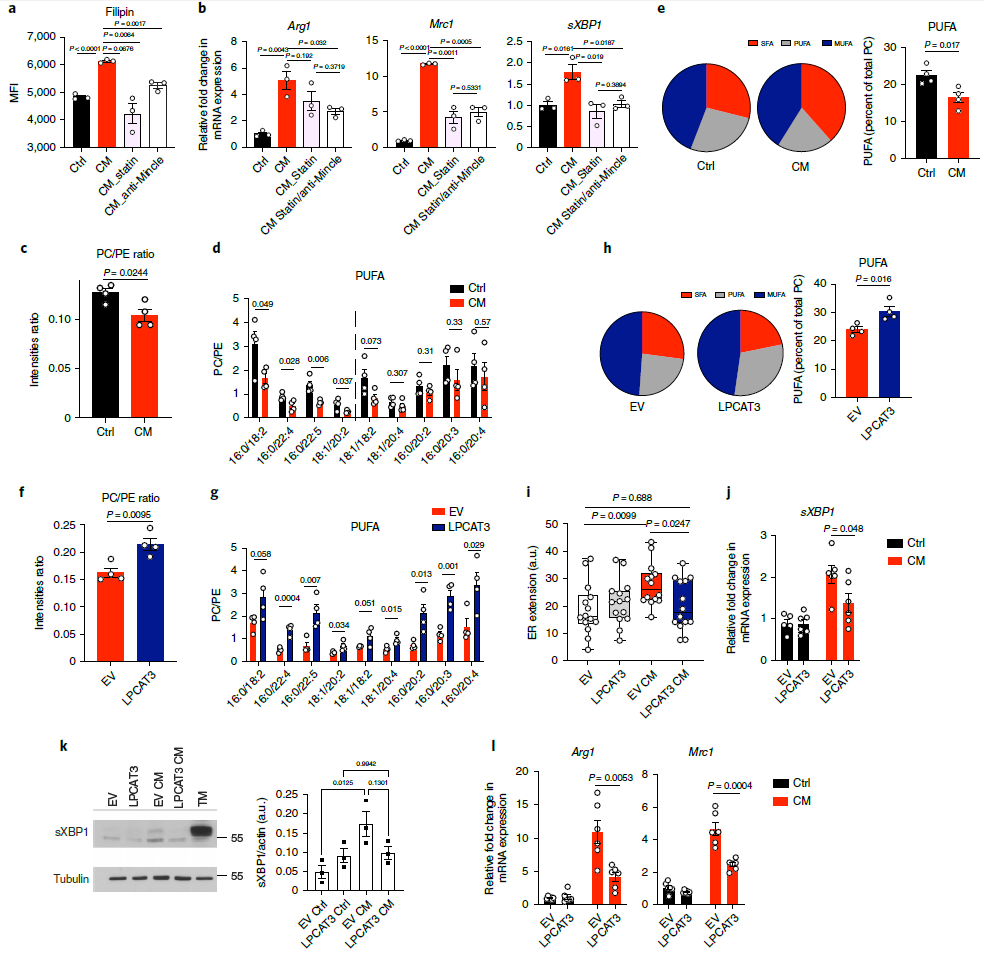
圖7 CM引起內(nèi)質(zhì)網(wǎng)膜脂質(zhì)重組和飽和
7、LXR激動劑通過LPCAT3控制巨噬細胞的腫瘤負荷
前人研究表明LPCAT3的表達受到LXR的調(diào)控,所以作者試圖用LXR激動劑GW3965誘導LPCAT3的表達,并研究這種處理是否可以用來定制TAMs的功能。GW3965在CM處理的BMDM中誘導LPCAT3(圖8a),并降低sXBP1產(chǎn)生,STAT3磷酸化,致瘤性基因的表達,部分緩解CM對CD8+T細胞的抑制性(圖8b-e)。此外,GW3965藥物處理可以減少腫瘤的體積和重量,表明GW3965可以抑制腫瘤進展,這和此前報道一致(圖8f-g)。為了進一步鑒定抗腫瘤反應(yīng)是否依賴于巨噬細胞中LPCAT3的表達,構(gòu)建了LPCAT3敲除鼠。結(jié)果顯示,在WT小鼠中,GW3965處理能顯著抑制腫瘤進展,而LPCAT3敲除后,這種抑制作用消失了(圖8h-l)。總的來說,與之前從xbp1缺陷小鼠獲得的數(shù)據(jù)一致,內(nèi)質(zhì)網(wǎng)應(yīng)激反應(yīng),特別是sXBP1的表達,對于TAMs的生存和致瘤極化是必需的,并且LXR介導的巨噬細胞LPCAT3誘導可以被利用來喚醒抗腫瘤反應(yīng)。
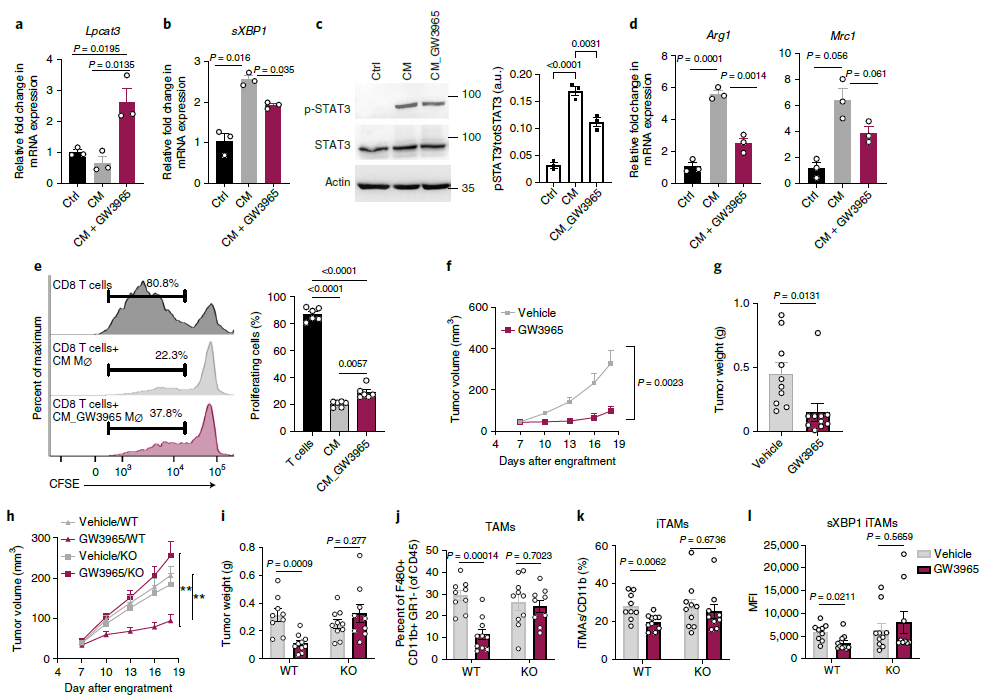
圖8 LXR激動劑以LPCAT3依賴的方式減少腫瘤負擔并阻礙TAM的生存