






長鏈非編碼RNA NR2F1-AS1通過調控NR2F1和ΔNp63 誘導乳腺癌肺轉移性休眠
彌散性腫瘤細胞通常在遠處器官中進入長期的休眠期,其特征是增殖減少但持續存活,直到轉移生長覺醒。然而,轉移性休眠和覺醒的調節機制在乳腺癌中仍然是未知的。本文中,作者發現乳腺癌干細胞樣細胞(BSCS)的上皮樣細胞和間充質樣細胞在肺中表現出不同程度的休眠和致瘤性,于是進一步探索調節轉移性休眠的機制。本文于2021年10月發表在《 Nature Communications》雜志上,IF=12.121。
本文技術路線:
主要結果如下:
許多研究報道了乳腺癌干細胞樣細胞(BCSCs)的異質性。早期在MCF10乳腺癌細胞系中發現了兩個BCSC亞群,其中的CD24?CD44med亞群(后面稱為P1),CD44染色較弱,但檢測為陽性。另一個CD24?CD44 high亞群(后面稱為P2),CD44染色更強。在小鼠中發現,與沒有CSC的細胞相比,P1和P2能顯著提高致瘤能力,但只有P1能誘導BCSC轉移。P1和富含P1細胞的轉移細胞系MCF10CA1a中有上皮標記物CDH1的表達;而P2為間充質樣亞群,能增強了體外遷移和侵襲特性。在含血清培養基中傳代多次后,P1細胞部分轉化為P2細胞,而在無血清培養基中則沒有出現這種情況。當細胞被注射到小鼠體內時,P2腫瘤往往顯示出更強的侵襲能力(Fig.1a)。
由于間充質樣P2細胞表現出更高的遷移、侵襲和播散能力,但與上皮樣P1細胞相比,肺轉移數目卻較少,作者試圖分析這些細胞的轉移過程。
富含P1細胞的轉移細胞系MCF10CA1a,轉移性較低的細胞系MCF10CA1h,以及P1和P2亞群(CA1h-P1和CA1h-P2)通過螢火蟲熒光素酶和GFP標記,再接種到小鼠中分析其在肺部的轉移情況,結果發現,與MCF10CA1a細胞系相比,MCF10CA1h細胞系出現更多的GFP標記,與CA1h-P1細胞相比,CA1h-P2細胞GFP標記更顯著(Fig. 1b, c)。然而,MCF10CA1h和CA1h-P2細胞在定殖初期不同時間點均有增殖活性(Fig. 1b, c)。小鼠生物熒光成像(BLI)的長期轉移監測也顯示初始播種期后肺內CA1h-P1穩定生長,而CA1h-P2細胞信號在18周后基本保持不變,但初始強度有所增強(Fig. 1d)。很多老鼠接種MCF10CA1a和CA1h-P1細胞最終在肺部發生轉移(Fig. 1e),肺表面可見明顯的腫瘤結節(Fig. 1f)和侵襲性腫瘤區域(Fig. 1g)。在20周內,MCF10CA1h和CA1h-P2保持為單細胞或小灶(Fig. 1g, h),導致腫瘤結節減少(Fig. 1f)。這些結果表明MCF10CA1h和CA1h-P2在肺部顯示出休眠表型。
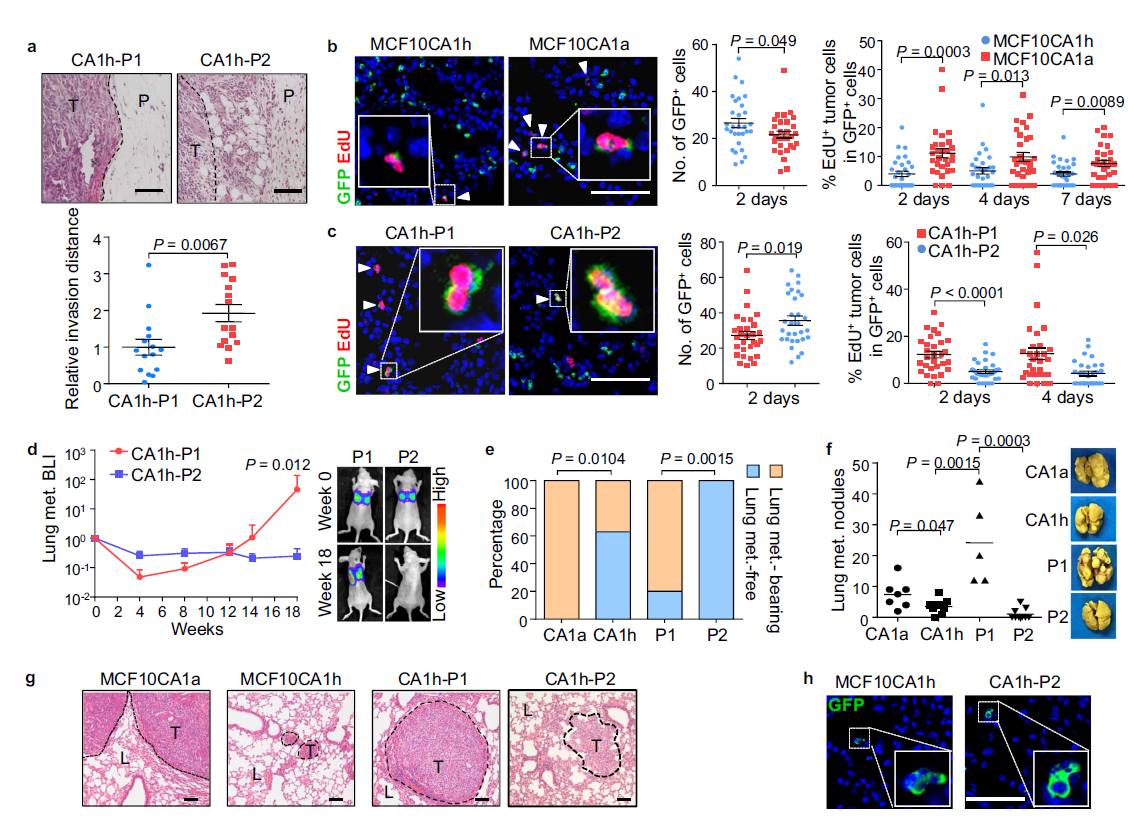
Fig1 P2 BCSC亞群在肺中顯示休眠表型
然后,作者將肺休眠細胞(MCF10CA1h和CA1h-P2)和肺轉移細胞(MCF10CA1a和CA1h-P1)以及在小鼠不能擴散到肺的MCF10AT進行測序比較。重點關注參與轉移性休眠的LncRNA,在肺休眠細胞中發現了18個上調和7個下調的LncRNA (Fig. 2a)。NAS1表達與腫瘤復發潛伏期相關,NR2F1-AS1(NAS1)位于蛋白編碼基因NR2F1的上游,但轉錄方向相反,屬于上調的LncRNA。NAS1的片段長度在顯示為3000bp左右(Fig. 2b)。進一步進行GSEA分析,顯示NAS1敲低導致CA1h-P2轉錄組向P1樣態轉變,在NAS1 敲除后雖然肺部GFP+癌細胞更少,但這些細胞更具有增殖能力(Fig. 2c),4個月后,肺部會發生轉移生長增強和甚至表現出更多的轉移結節(Fig. 2d, e)。相反,過表達NAS1雖然在MCF10AT和CA1h-P1中增強了初始癌細胞在肺部的數量,但削弱了其增殖能力(Fig. 2f),導致肺部單個腫瘤細胞持續存在,最終導致轉移性腫瘤結節減少(Fig. 2g,h)。將乳腺癌細胞的肺轉移亞系MDA-MB-231原位注射到小鼠的乳腺脂肪中。后續分析顯示NAS1增強腫瘤侵襲能力(Fig. 2i),但導致更多獨立的腫瘤細胞并造成更少的轉移性結節(Fig. 2 j, k)。這些數據表明,NAS1促進腫瘤細胞的轉移,但抑制轉移性產物的擴散,導致乳腺癌細胞在肺部的休眠。

Fig. 2 NR2F1-AS1 (NAS1)促進乳腺癌的轉移性休眠
3.NAS1促進EMT和侵襲,但抑制癌細胞的腫瘤啟動能力
為了分析為什么NAS1會促進癌細胞生長,但抑制肺轉移,作者首先分析了癌細胞的EMT狀態和侵襲性。從細胞形態和各種EMT標記物的表達可以看出(Fig. 3a),在CA1h-P2 CSC亞群中,NAS1敲低可部分誘導上皮間充質轉化(MET)。此外,作者還觀察到,在CA1h-P2細胞中,NAS1敲低導致CD44下調,而在MCF10AT細胞中ALDH上調,而過表達NAS1能促進ALDH+細胞向cd44間質轉化CSCs((Fig. 3b–e),證實了NAS1促進上皮-間質轉化的作用。 轉移性生長需要擴散的癌細胞產生新的腫瘤的能力。因此,作者接下來進一步評估NAS1在腫瘤啟動中的作用。NAS1敲低顯著增加CA1h-P2形成腫瘤的能力(Fig. 3f)。此外,腫瘤發生的限制性稀釋試驗顯示,NAS1敲低極大地增加了體內腫瘤的發生和CA1h-P2引起的的腫瘤生長(Fig. 3g, h)。相反,在MCF10AT中過表達NAS1將會阻遏腫瘤的形成(Fig. 3i)。此外,作者發現在MCF10CA1a中過表達NAS1會降低了小鼠細胞的致瘤能力(Fig. 3j),,并降低原發腫瘤切除后的腫瘤復發率(Fig.3k)。總的來說,NAS1促進了腫瘤擴散和擴散所必需的癌細胞特征,包括EMT、遷移和侵襲,但抑制了啟動新腫瘤生長的能力,從而導致了傳播后休眠。

Fig3. NAS1促進EMT但抑制乳腺癌細胞的致瘤性
4. NAS1通過上調NR2F1來促進休眠
接下來,作者探討了NAS1在轉移調控中的分子機制。重點關注了位于NAS1附近的NR2F1基因,該基因編碼一個促進腫瘤休眠的轉錄因子。作者發現NR2F1在不同轉移能力的MCF10細胞中存在差異表達,NAS1能上調NR2F1的表達(Fig. 4a) 。此外,NR2F1過表達表現出類似NAS1的作用,能誘導細胞增殖(Fig. 4b),并在多個細胞系中抑制腫瘤球的形成(Fig. 4c)。為了進一步驗證NR2F1是否作用于NAS1下游調控轉移性休眠,將NR2F1在NAS1敲低的CA1h-P2細胞中過表達。NR2F1有效地恢復了這些細胞的間充質樣表型(Fig. 4d)。此外,NR2F1降低了因NAS1敲低而增強的腫瘤形成能力(Fig. 4e, f)。更重要的是,當CA1h-P2細胞接種到小鼠體內,NR2F1過表達消除了NAS1敲除的作用,抑制了腫瘤細胞增殖(圖4g),減少了肺轉移瘤的生長和腫瘤結節的形成。這些數據提示,NAS1通過調節NR2F1促進乳腺癌轉移性休眠。
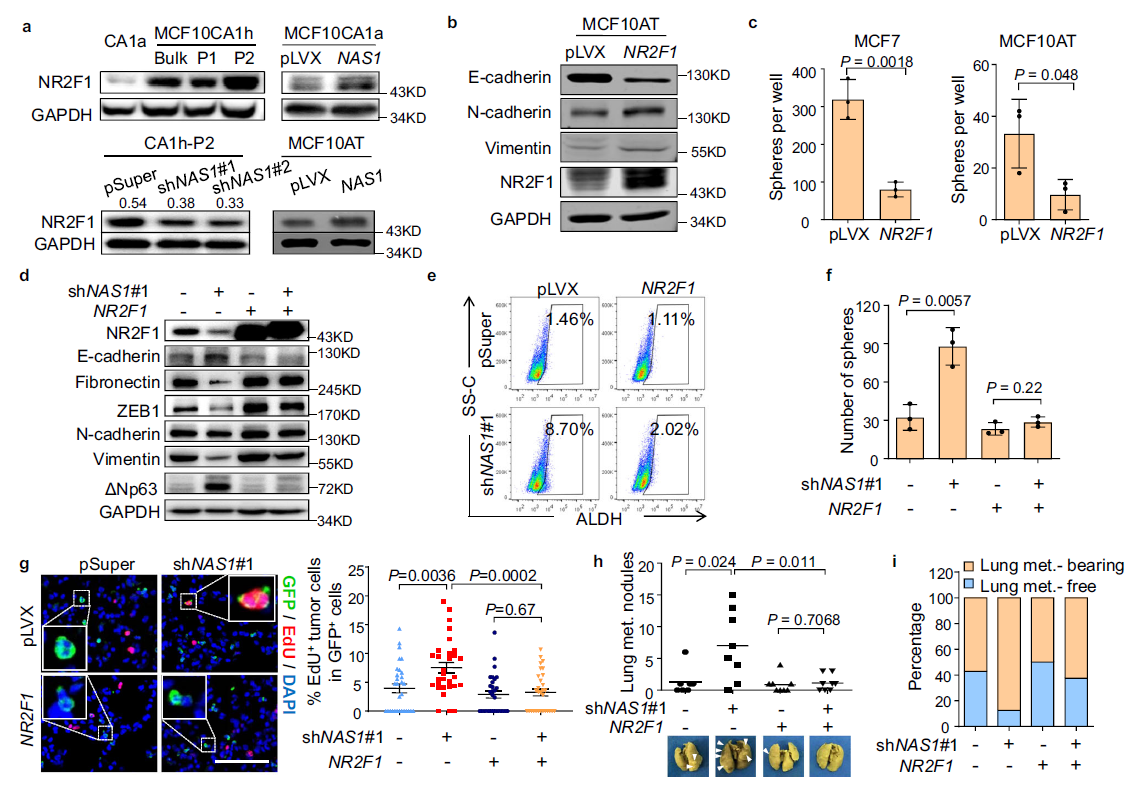
Fig4. NAS1通過NR2F1抑制乳腺癌肺轉移
5.NAS1和PTBP1共同增強NR2F1-5'UTR的IRES活性
為了進一步研究NAS1如何調控NR2F1的表達,作者對NAS1進行RNA下拉實驗,并進行質譜分析。將PTBP1作為結合蛋白,通過 WB實驗發現 PTBP1能拉下NAS1 (Fig. 5a)。RNA免疫沉淀法也檢測到PTBP1的免疫沉淀中含有NAS1, RNA(RIP)分析,進一步證明NAS1與PTBP1 的結合(Fig. 5b)。
接下來,作者分析了PTBP1在NR2F1表達中的作用。在MCF10AT中,過表達PTBP1可上調NR2F1,而在MCF10CA1h中,對 PTBP1進行干擾可明顯降低NR2F1的蛋白水平,提示PTBP1參與了NAS1對NR2F1的調控。事實上,在MCF10AT中,敲除PTBP1以及過表達NAS1完全阻斷了NR2F1通過NAS1表達;而在CA1h-P2中,PTBP1與NAS1敲除后,NR2F1蛋白水平恢復(Fig. 5c),證實了PTBP1介導了NAS1在NR2F1調控中的作用。
進一步分析PTBP1如何調控NR2F1的表達,在排除了PTBP1調控NR2F1啟動子活性的可能性之后,通過RIP和融合蛋白免疫沉淀試驗均發現NR2F1 mRNA與PTBP1結合(Fig. 5e),顯示了PTBP1蛋白、NAS1 RNA和NR2F1 mRNA之間的相互作用。此外,作者還發現NAS1敲低可破壞PTBP1和NR2F1 mRNA的結合(Fig. 5f)。而抑制PTBP1后,拉下的NAS1中NR2F1 mRNA的下調無明顯變化(Fig. 5g )。因此,這些數據表明NAS1促進PTBP1和NR2F1 mRNA的結合。
接下來,作者分析了NAS1和PTBP1如何調控NR2F1 mRNA。由于在NR2F1 mRNA的5'UTR中發現了多個多嘧啶區域,推測PTBP1和NAS1可能促進了NR2F1- 5'UTR的IRES功能。為了驗證這一假設,作者構建了IRES活性報告質粒(Fig. 5h)以及NR2F1-5'UTR段(Fig. 5h),RES報告分析顯示UTR具有輕微的啟動翻譯的活性,而5 'UTR-PT表現出更強的IRES活性。相反,5'UTR-GC完全沒有IRES活性(圖5i),接下來,作者通過pGL3報告基因評估了這些區域的潛在啟動子活性,結果發現雖然5′UTR 和5′UTR-GC檢測到輕微的啟動活性,但5′UTR-PT沒有啟動轉錄的能力,表明NR2R1 5'UTR的多嘧啶區具有先天的IRES活性,而下游富含gc的序列的存在阻礙了這種活性的mRNA翻譯。
有趣的是,通過蛋白-RNA,RNA-RNA相互作用預測工具(catRAPID61和IntaRNA62-65)進行分析,發現PTBP1和NAS1分別與NR2F1-5 '的多嘧啶和gc富集區域結合(Fig. 5j)。通過RIP和pull down實驗對NR2F1 mRNA不同區域的進行RIP和RNA下拉分析,證實了NAS1 RNA優先結合5 'utr-gc(Fig. 5k), PTBP1只與5′UTR-P特異結合(Fig. 5l)。而且抑制PTBP1導致了5'UTR-PT IRES活性受到抑制(Fig. 5m)。反之,NAS1過表達可增強IRES 5'UTR的活性,但這種調節依賴于GC富集。5'UTR-PT較強的IRES活性不受NAS1的影響(圖5n)。由此可見,PTBP1與NR2F1-5 ' utr的多嘧啶區域結合啟動IRES活性,但NR2F1的翻譯過程受到下游富gc區域的抑制。這種抑制可以通過與NAS1的結合而解除,可能是通過重塑在這個富含gc的區域形成的RNA結構(Fig. 5o)。
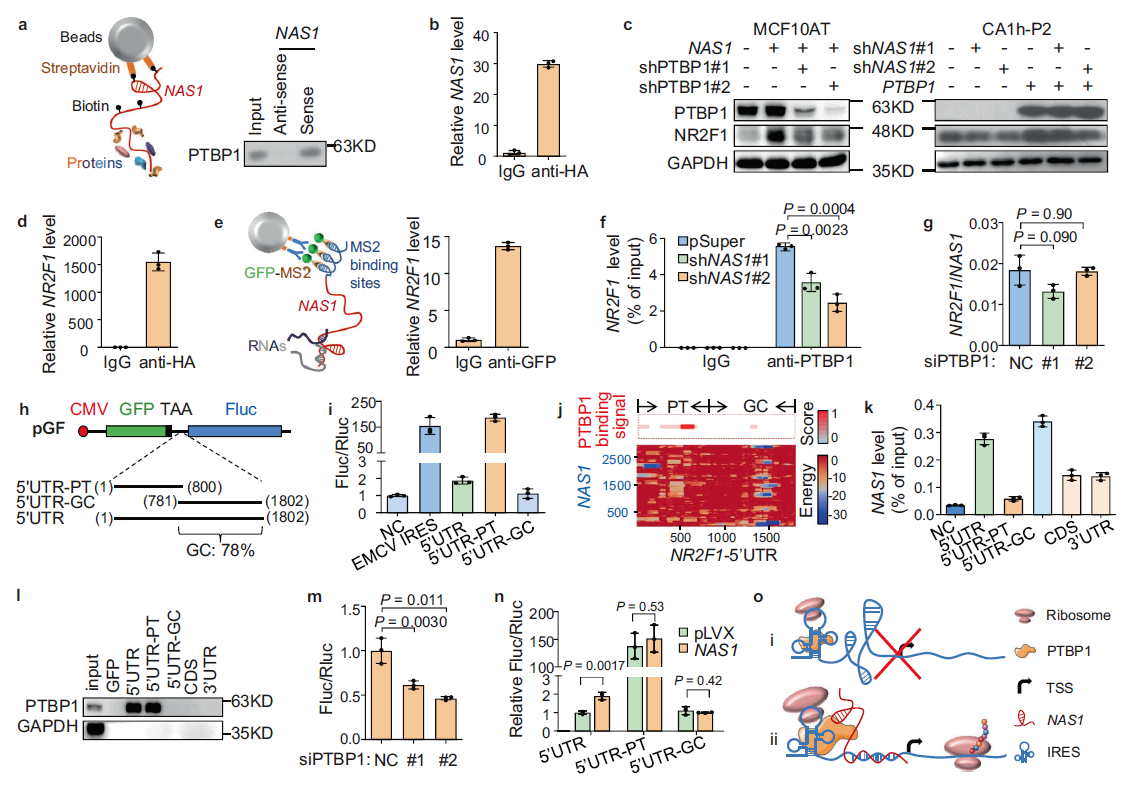
Fig 5 NAS1和PTBP1共同增強NR2F1-5'UTR的IRES功能
6. NR2F1抑制ΔNp63的表達
為進一步分析NAS1-NR2F1促進腫瘤細胞休眠的下游分子事件,通過對NAS1敲低或NR2F1過表達的癌細胞進行RNA測序分析。發現了miR-205和發生了miR-205的轉錄調控因子TP63基因變異的ΔNp63。已有報道闡明了miR-205通過靶向ZEB1和SIP168抑制腫瘤細胞EMT。
作者分析發現,miR-205和ΔNp63的表達均被NAS1和NR2F1顯著抑制(Fig. 6a),,這一點被qPCR和WB檢測進一步證實(Fig.6b–d)。
因此,作者對ΔNp63在NAS1誘導EMT和抑制腫瘤進行研究。在CA1h-P2細胞中,NAS1敲低的同時抑制ΔNp63,能逆轉MET表型,恢復間質形態和標志物表達的(Fig. 6e)。而干擾ΔNp63也有效抑制了因NAS1基因敲低而造成腫瘤球形成能力(Fig. 6f)。GSEA分析發現NR2F1對ΔNp63的下游基因有抑制作用(Fig. 6g),。ChIP-qPCR分析顯示NR2F1在ΔNp63啟動子上結合(Fig.6h)。綜上所述,這些數據表明NR2F1通過抑制ΔNp63的表達介導了NAS1的休眠促進功能。處理時,在CA1h-P2細胞中,抑制miR-205恢復了NAS1敲低造成的間葉細胞表型,NAS1過表達MCF10AT將會誘導細胞上皮恢復(Fig. 6i)。因此,miR-205作用于NAS1-NR2F1 -ΔNp63信號軸下游調控細胞EMT。
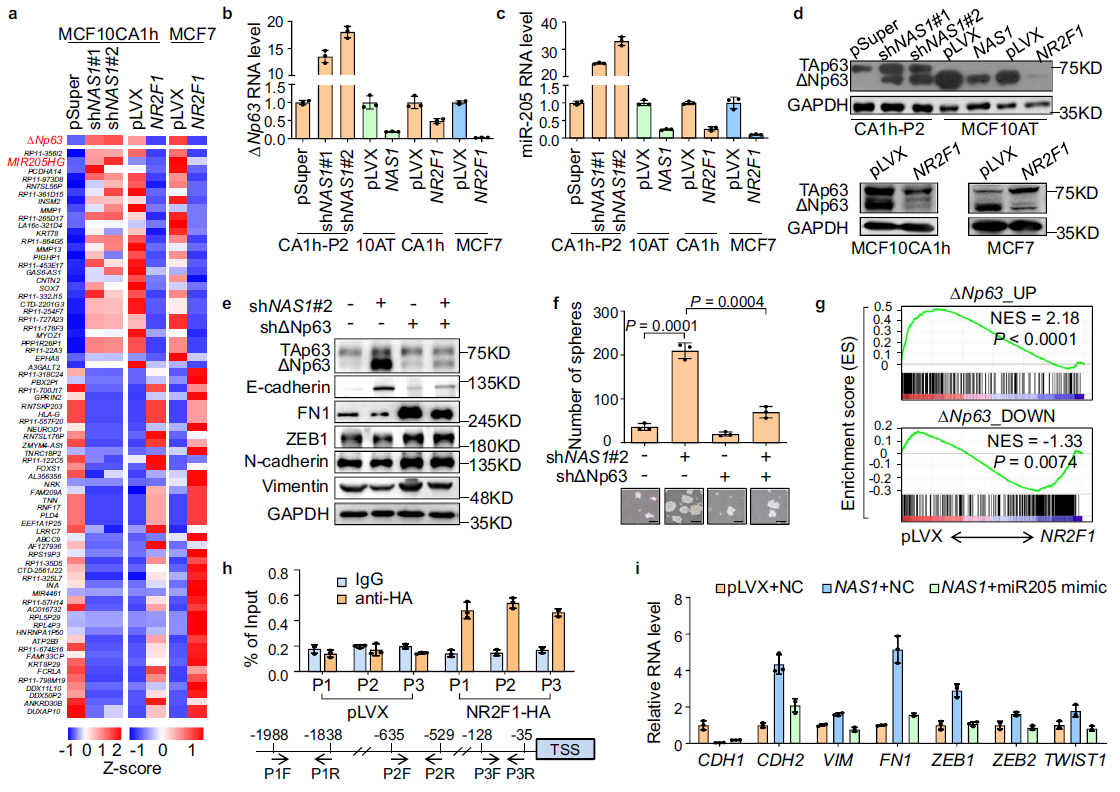
Fig 6 NR2F1通過抑制ΔNp63轉錄介導NAS1在休眠中的作用
7. 在乳腺腫瘤中,NAS1與NR2F1、EMT標記物和轉移減少相關
最后,作者評估了NAS1的表達在乳腺腫瘤樣本中的臨床意義。首先,在乳腺癌患者的腫瘤樣本中發現NAS1 RNA與NR2F1蛋白水平呈正相關(Fig. 7a), 驗證了NAS1在調控中的作用NR2F1。然后,通過對RNA測序數據的分析,發現了NAS1與大量emt相關基因顯著相關(Fig. 7b)。在乳腺癌陰性患者中,NAS1也與之前報道的M-BCSC和EMT基因標記的富集以及E-BCSC標記的缺失呈正相關(Fig. 7c)。其次,從齊魯醫院收集的89例乳腺癌樣本分析發現腫瘤NAS1高表達與較低的轉移和腫瘤復發風險相關(Fig. 7d, e)。
同時,Kaplan-Meier Plotter臨床數據庫74的分析也顯示了NAS1改善了乳腺癌復發情況。通過轉錄組數據分析也發現敲低NAS1或過表達NR2F1都會促進 NAS1基因表達,與加速或抑制腫瘤轉移相關(Fig. 7f, g)。最后,與正常乳腺組織相比,乳腺腫瘤中NAS1的下調(Fig. 10g),驗證了NAS1抑制致瘤性的作用。
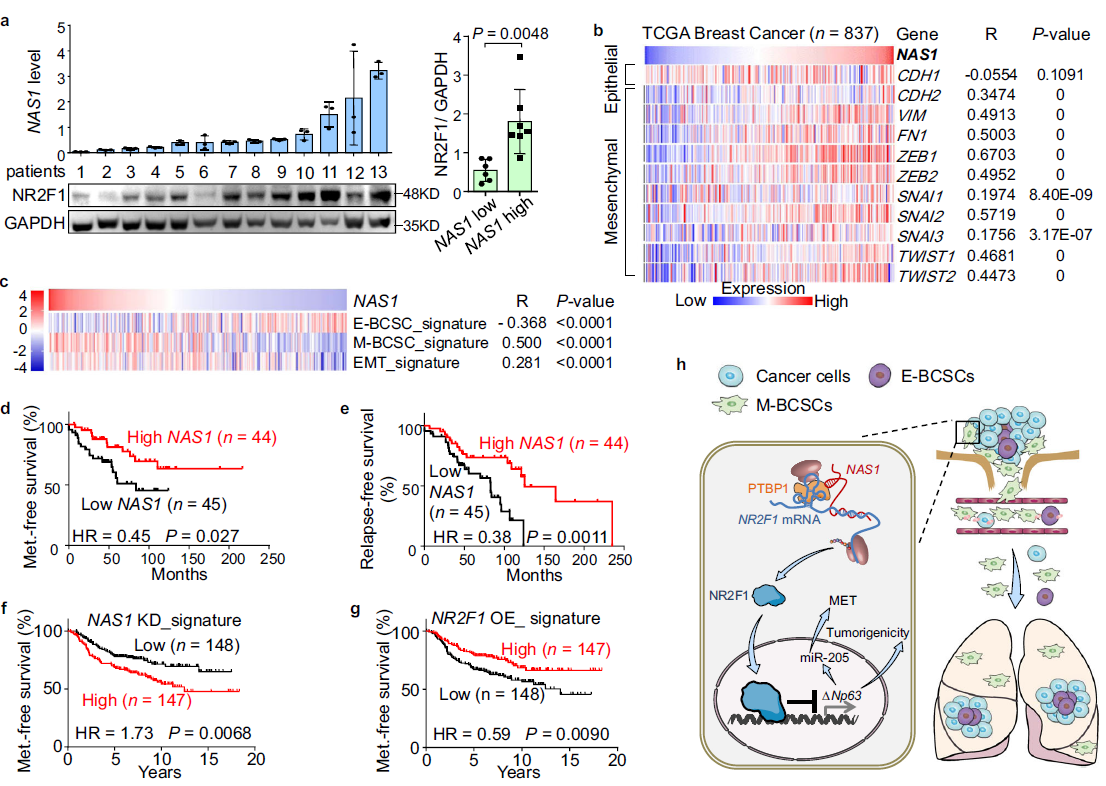
Fig 7 NAS1的表達在乳腺腫瘤中及其臨床意義
本研究闡明了間充質腫瘤細胞在轉移過程中的休眠機制。
1. Liu, Y., et al., Author Correction: Long non-coding RNA NR2F1-AS1 induces breast cancer lung metastatic dormancy by regulating NR2F1 and DeltaNp63. Nat Commun, 2021. 12(1): p. 5973.