






LncRNA-HGBC通過調節miR-502-3p/SET/AKT通路促進膽囊癌發展
LncRNA在很多癌癥的發生和發展中起著重要作用,但在膽囊癌(GBC)中,很多新的LncRNA如何對疾病進行調節需進一步研究。本文中,作者通過芯片篩選出膽囊癌(GBC)患者中膽囊惡性腫瘤與周圍良性組織微陣列中差異表達的LncRNA和mRNA,并闡明其對GBC細胞增殖、遷移、侵襲的作用機制。
本文技術路線如下:

主要結果如下:
1 、鑒定出LncRNA-HGBC,并發現LncRNA-HGBC表達水平與GBC預后相關
為找出膽囊惡性腫瘤與周圍良性組織中差異表達的LncRNA和mRNA,通過芯片技術篩選出一些候選LncRNA,并發現其中的LncRNA-HGBC(LncRNA Highly expressed in GBC)在GBC腫瘤細胞中表達量顯著高于周圍良性組織,且LncRNA-HGBC高表達的患者總生存率(OS)顯著低于LncRNA-HGBC低表達的患者(Fig 1A and B),揭示了LncRNA-HGBC在膽囊癌腫瘤細胞轉移中起積極作用。隨后,作者通過RACE快速擴增得到LncRNA-HGBC的5′端及3′端序列(Fig 1C),再通過PCR 擴增GBC全長序列,Northern雜交進一步證實在GBC細胞系中LncRNA HGBC的RNA全長約為2 kb(Fig 1D)。為確定LncRNA-HGBC所在位置,作者分離了NOZ細胞的細胞核和細胞質部分,進行了qRT-PCR檢測,初步判斷LncRNA-HGBC主要定位在細胞質中,與原位雜交(FISH)結果一致(Fig 1E)。通過體外翻譯實驗,發現LncRNA-HGBC并未參與任何蛋白質的表達(Fig 1F),表明LncRNA-HGBC是一個不編碼蛋白質的長鏈RNA。
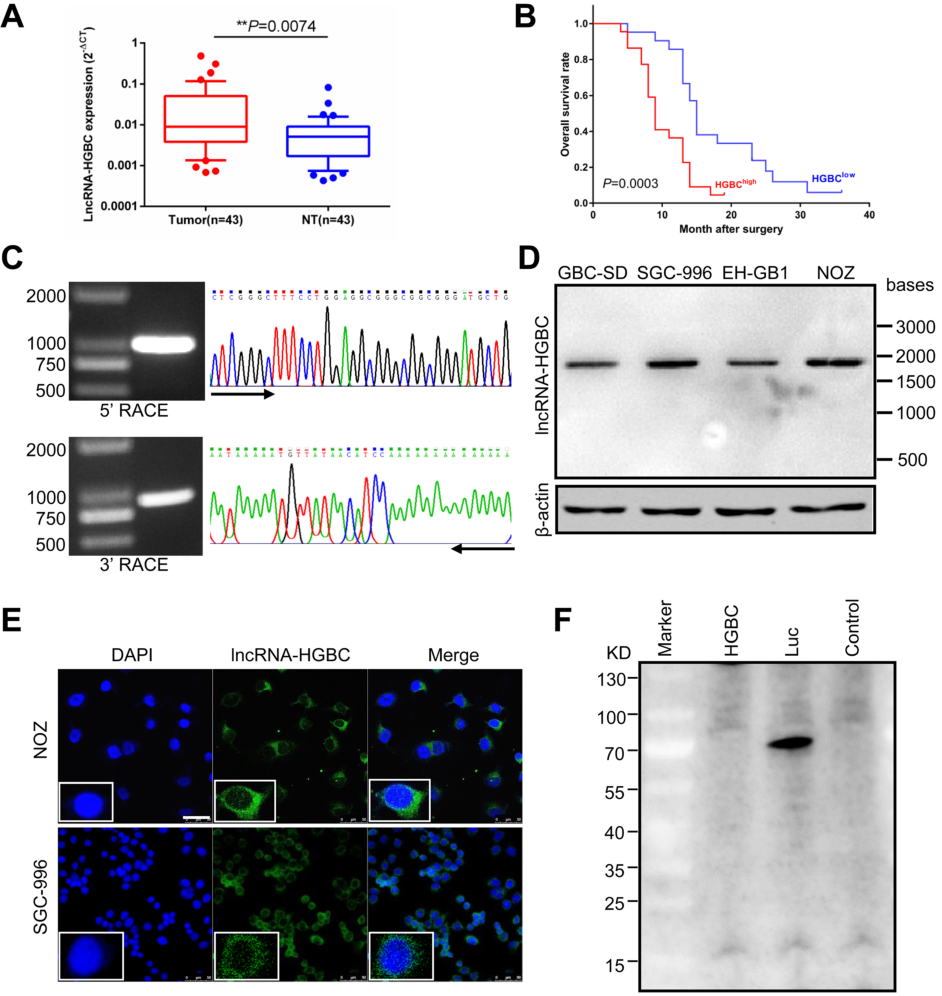
Figure 1 影響GBC進展的LncRNA的鑒定與表達分析
2、LncRNA-HGBC能在體內外促進GBC細胞增殖
為確定LncRNA-HGBC對GBC細胞增殖及對腫瘤發生的影響,研究中檢測了LncRNA-HGBC在四種GBC細胞系(NOZ、GBC-SD、SGC-996、EH-GB1)中的表達情況,結果發現NOZ和SGC-996細胞系中LncRNA-HGBC的表達量高于GBC-SD和EH-GB1細胞系(Fig 2A)。在LncRNA-HGBC高表達的NOZ和SGC-996細胞系中通過干擾LncRNA-HGBC(Fig 2B),發現任意干擾兩個區域都會使細胞增殖顯著降低(Fig 2C),于此同時,干擾后菌落數量也顯著減少,兩種細胞系形成菌落的能力降低(Fig 2D)。此外,為了確定LncRNA-HGBC在體內對GBC細胞生長的影響,在小鼠中比較注射敲除了LncRNA HGBC的NOZ細胞和注射未經敲除的NOZ細胞時小鼠之間腫瘤差異,結果發現,前者較后者的腫瘤體積和腫瘤重量降低30%(Fig 2E)。通過免疫組化檢測增殖細胞核抗原(PCNA),觀察細胞增殖情況,也得到了相同的結論,與對照組比較,LncRNA hGBC水平降低會引起細胞增殖水平下降(Fig 2F)。另一方面,在GBC-SD 和 EH-GB1細胞系中,通過建立LncRNA hGBC過表達的發現過表達細胞系中細胞的增殖速度明顯提高(Fig 2G 、2H、2I),此外,過表達細胞系中腫瘤的體積和重量是對照組的2.5倍(Fig 2J),通過IHC染色發現過表達細胞系異種移植瘤組織中增殖細胞核抗原(PCNA)上調(Fig 2K)。
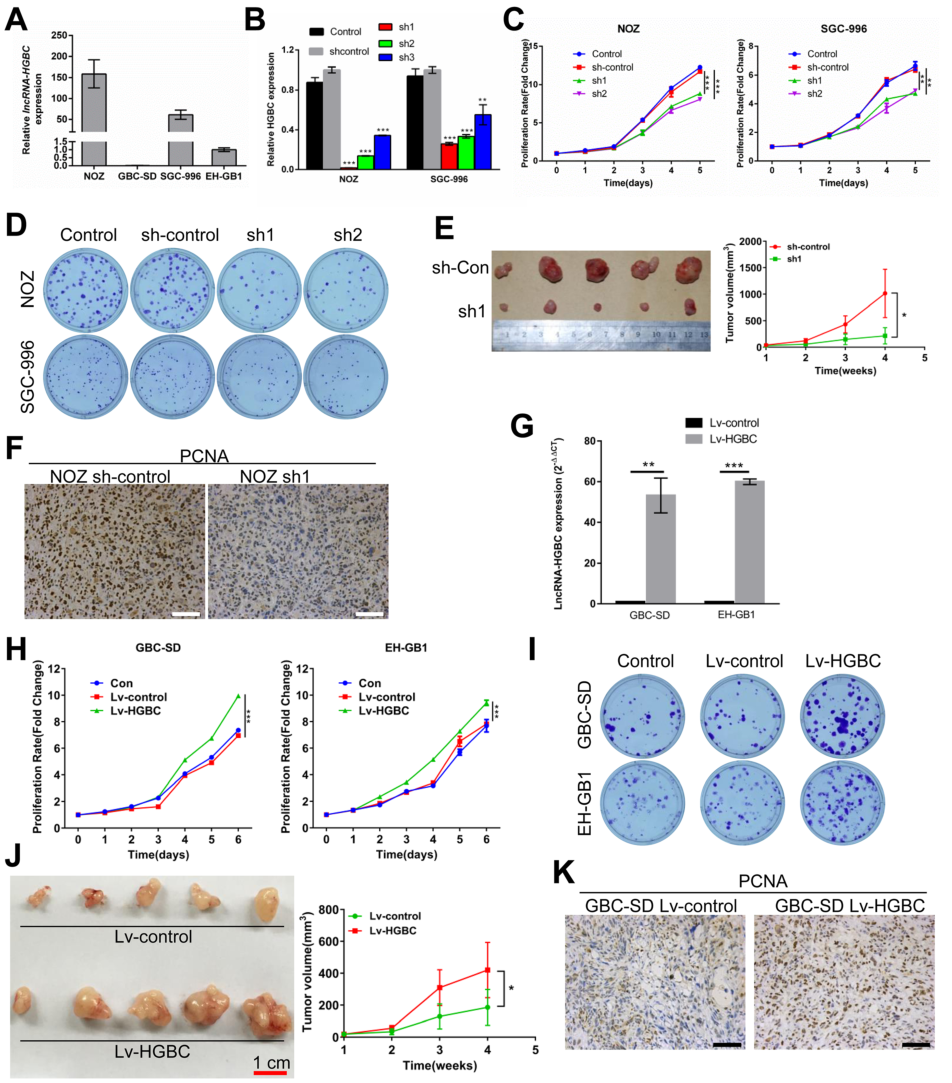
Figure 2 LncRNA-HGBC促進GBC細胞增殖和腫瘤生長
3、LncRNA -HGBC促進GBC細胞進行上皮細胞-間充質轉化(EMT)和腫瘤轉移
為了判斷LncRNA –HGBC表達水平與淋巴結細胞遷移之間的關系,研究中首先提出了LncRNA –HGBC促進GBC細胞遷移的假設,并進行驗證。通過transwell和基質膠侵襲實驗,發現在NOZ 和 SGC-996 細胞系中,敲除LncRNA -HGBC可顯著減少約30-40%的細胞遷移和侵襲(Fig 3A,3B)。反之,在GBC-SD 和 EH-GB1細胞系中細胞,對LncRNA- HGBC進行過表達,發現在GBC細胞遷移和侵襲能力增加20-50%(Fig 3C,3D)。EMT(上皮細胞-間充質轉化)在很多腫瘤細胞遷移和侵襲中非常重要,為了評估EMT是否參與GBC侵襲,本研究中檢測了兩種EMT特異性標記物,分別是波形蛋白和N-鈣粘蛋白。作者在NOZ 和SGC-996 cells細胞系中,敲除LncRNA –HGBC發現波形蛋白和N-鈣粘蛋白的表達量降低(Fig 3E),反之,在GBC-SD 和 EH-GB1細胞系中,對LncRNA- HGBC進行過表達,發現波形蛋白和N-鈣粘蛋白的表達量升高(Fig 3F),說明LncRNA- HGBC可能引起EMT的發生。為進一步驗證,作者將NOZ細胞注射到小鼠脾臟中建立了裸鼠肝轉移瘤模型,對照組的6只小鼠中有5只(5/6)在移植6周后顯示熒光素酶信號增強,并在肝內發生轉移,而注射 干擾LncRNA-HGBC后只小鼠中只有3只小鼠出現肝內轉移。在肝臟中,對 LncRNA-HGBC 進行干擾的小鼠中轉移灶較對照組降低10%(Fig 3G,3H)。
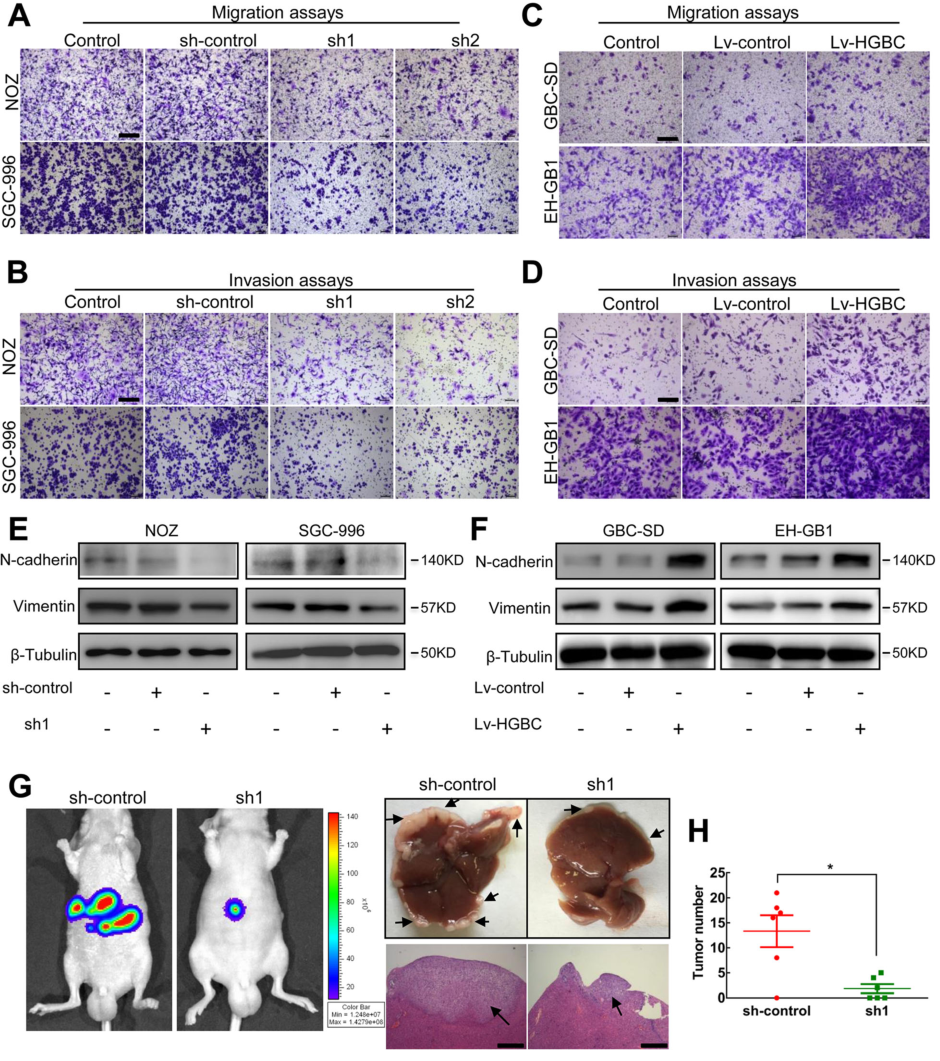
Figure 3 LncRNA-HGBC增強GBC細胞的侵襲能力
4、 HuR與LncRNA-HGBC特異結合并維持LncRNA-HGBC穩定表達
為探索LncRNA-HGBC與是否與GBC細胞存在潛在蛋白,通過pull-down實驗,發現LncRNA-HGBC在37kd處存在與蛋白質的互作(Fig 4A),進一步通過 Western免疫印跡證實與LncRNA-HGBC結合的蛋白為HuR(Fig 4B),通過RNA結合蛋白免疫沉淀(RIP)驗證LncRNA-HGBC與HuR的互作,與富含HuR結合位點的陽性對照EIF4E mRNA相比,通過RNA結合蛋白免疫沉淀(RIP)實驗證驗了LncRNA HGBC和HuR之間的特異性相互作用(Fig 4C)。為了進一步確定LncRNA HGBC中的哪個特定區域與HuR結合有關,作者構建了4個不同的LncRNA -HGBC缺失片段(Fig 4D),通過pull-down實驗之后再進行Western免疫印跡確定了LncRNA -HGBC的1759–1906nt區域與HuR特異結合(Fig 4E)。為了闡明HuR是否影響LncRNA-HGBC在GBC細胞中的穩定性,通過兩次HuR敲除實驗均發現LncRNA-HGBC在NOZ細胞系中的表達量降低(Fig 4F);為進一步探索 LncRNA-HGBC表達量的降低是不是由于 LncRNA-HGBC衰退所致,通過阻斷RNA的轉錄,觀察30h內 LncRNA-HGBC表達量的變化,結果發現,隨著HuR的逐步消耗,LncRNA-HGBC水平的半衰期從18小時降低到5小時(Fig 4G),這一系列實驗表明HuR與LncRNA HGBC相互作用并維持LncRNA HGBC穩定表達。
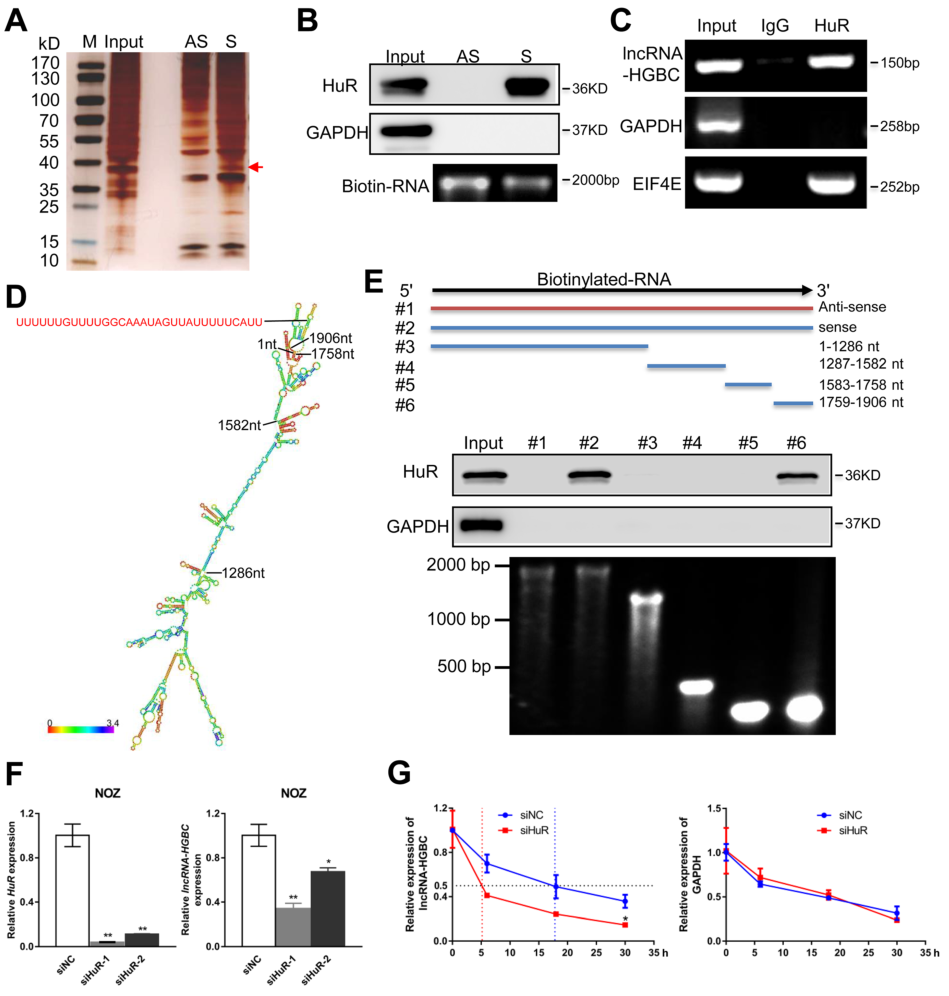
Figure 4 HuR與LncRNA-HGBC特異結合并有助于維持LncRNA-HGBC穩定表達
5、 LncRNA-HGBC作為一種競爭性內源性RNA抑制miR-502-3p表達
前期的研究已經證實LncRNA-HGBC定位在細胞質上,作者初步假設LncRNA -HGBC可能作為miRNA海綿影響miRNA的表達,在GBC的進展中發揮作用。為了找出與LncRNA-miRNA相互作用的miRNA, 作者將LncRNA-HGBC全長序列克隆到pmirGLO雙熒光素酶報告載體中,將6個與腫瘤抑制相關的候選miRNA通過雙熒光素酶實驗進行分析,發現其中的miR-502-3p和miR-618可抑制LncRNA-HGBC的熒光素酶活性(Fig 5A),說明LncRNA-HGBC可能與miR-502-3p、miR-618相互作用。作者根據先前的miRNA微陣列結果發現,與鄰近的非腫瘤組織相比,在GBC組織中miR-502-3p表達量確實下調了。當LncRNA-HGBC中1176 - 1183nt之間的序列被敲除后將不能與miR-502-3p結合,熒光活性將不會再被抑制(Fig 5B)。AGO2蛋白可以在轉錄后調控調節miRNA豐度,內源性Ago2的減少會降低成熟miRNA的表達和活性。進一步進行免疫共沉淀實驗,發現AGO2抗體復合物在LncRNA-HGBC和miR-502-3p中特異富集,表明miR-502-3p是一個真正的靶向LncRNA-HGBC的miRNA(Fig 5C)。為進一步確定LncRNA-HGBC是否與miR-502-3p結合,作者通過MS2-RIP找到與LncRNA-HGBC結合的 miRNA,結果再一次證實了miR-502-3p與LncRNA-HGBC特異性結合(Fig 5D),而且miR-502-3p結合位點的突變也使miR-502-3p與LncRNA-HGBC之間的結合不再成立,也驗證了LncRNA-HGBC和miR-502-3p之間的直接結合。為了進一步闡明LncRNA-HGBC和miR-502-3p之間基因表達的調控關系,評估了不同lncRNA-HGBC表達水平對細胞中的miR-502-3p水平的影響。結果發現,在 NOZ細胞系中,敲低LncRNA-HGBC 會導致miR-502-3p表達量上調了60%(Fig 5E左圖),在EH-GB1細胞系中,LncRNA-HGBC過表達后miR-502-3p的表達被抑制,表達量下降了50%(Fig 5E右圖), 但對miR-502-3p進行敲除或過表達時LncRNA-HGBC的表達量沒有變化,說明LncRNA-HGBC作為海綿會抑制miR-502-3p表達,但不能誘導其降解。為了檢測LncRNA HGBC的活性是否取決于其與miR-502-3p的結合,在異位表達野生型LncRNA HGBC和結合突變型HGBC-MUT后進行cck-8和Transwell分析,結果發現,LncRNA-HGBC(而非HGBC-MT)的表達可顯著促進細胞遷移、侵襲和增殖(Fig 5F-G),由此可見,LncRNA-HGBC與miR-502-3p能結合,并可作miR-502-3p的海綿抑制其表達。
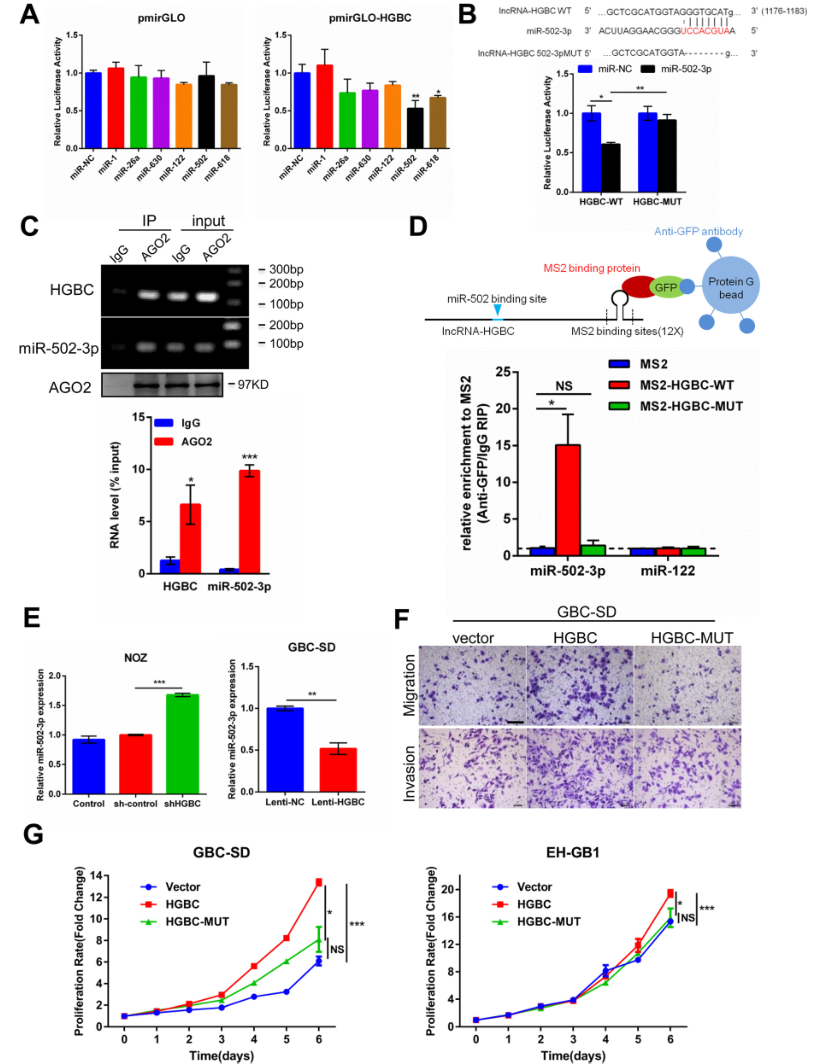
Figure 5 LncRNA-HGBC作為一種競爭性內源性RNA能直接與miR-502-3p結合
6、LncRNA-HGBC通過競爭性結合miR-502-3p調節SET的表達
miR-502-3p可以靶向多種蛋白質,并在癌癥發展過程中發揮重要,作者通過芯片數據分析發現SET在GBC組織中也上調,那么miR-502-3p是否對SET進行調節需進一步探索。作者將含有SET 3′UTR的熒光素酶報告載體轉染到293T細胞中,然后在轉染miR-502-3p模擬物時評估熒光素酶活性,發現miR-502-3p顯著降低了SET報告載體的熒光素酶活性此外,在GBC細胞系中,對miR-502-3p進行過表達或敲除之后,SET隨之變化,說明SET是miR-502-3p的直接靶點。
由于LncRNA-HGBC和SET 3′UTR都與miR-502-3p結合,于是進一步研究LncRNA HGBC是否調節了GBC細胞中miR-502-3p,并對SET進行抑制。將pmirGLO-SET熒光素酶報告載體與LncRNA-HGBC過表達質粒進行共轉染,過表達miR-502-3p將會抑制SET報告載體的熒光素酶活性,但過表達cRNA-HGBC將會誘導SET報告載體的熒光素酶活性(Fig 6A)。當miR-502-3p和LncRNA-HGBC同時被添加時,熒光活性恢復到對照水平,這就意味著LncRNA- HGBC能夠通過與miR-502-3p結合,進而上調SET。進一步研究SET 如何控制蛋白表達,在內源性LncRNA HGBC 高表達的 NOZ細胞系中進行LncRNA-HGBC干擾和miR-502-3p抑制,結果發現LncRNA-HGBC的敲除抑制了SET的表達,miR-502-3p抑制劑的加入反而促進了SET的表達(Fig 6B,左圖)。然而,在內源性LncRNA HGBC 低表達的EH-GB1細胞系中,過表達LncRNA-HGBC誘導SET表達,此時加入miR-502-3p,SET表達量將會降低至對照水平(6B,右圖)。在NOZ細胞細胞系中,對LncRNA HGBC進行干擾將會抑制細胞增殖、遷移和侵襲能力,而miR-502-3p的加入逆轉了這種局勢(Fig 6CandD) 。
然而,在EHGB1細胞系中,過表達miR-502-3p延緩了細胞的生長、遷移和侵襲(Fig 6C and E)。為檢測LncRNA HGBC是否誘導SET表達是否取決于LncRNA HGBC與miR-502-3p的結合,在GBC-SD和EH-GB1兩個細胞系中,通過過表達LncRNA-HGBC發現mRNA和蛋白質水平上SET表達上調,LncRNA HGBC的過度表達導致SET表達增加,而LncRNA HGBC的miR-502-3p結合位點突變卻未能誘導SET表達(Fig 6FandG),通過IHC發現,腫瘤異種移植模型中在LncRNA-HGBC耗盡之后SET的表達量也隨之降低,過表達LncRNA-HGBC的腫瘤異種移植模型中LncRNA-HGBC表達量顯著增加,說明LncRNA-HGBC和SET之間存在共表達,LncRNA-HGBC通過競爭性結合miR-502-3p調節SET的表達。

Figure 6 LncRNA-HGBC通過競爭性結合miR-502-3p調節SET的表達
7、AKT是SET的下游效應物、lncRNA與miR-502-3p、SET和p-AKT的關系
接下來對LncRNA-HGBC和SET對影響GBC轉移的分子機制進行研究。研究發現對SET進行敲除后GBC細胞的增殖和侵襲能力明顯降低,已有研究表明SET在很多癌癥中是通過激活AKT提高癌細胞活性,那么LncRNA-HGBC-miR-502-3p-SET-AKT是否作為一個完整的信號通路介導GBC發生和轉移的需要進一步驗證。
作者首先檢測了AKT激酶的活性,在NOZ和SGC-996細胞系中,通過WB實驗發現了敲除LncRNA -HGBC顯著抑制了AKT的活性(Fig 7A);在GBC-SD和EH-GB1細胞系中,過表達LncRNA-HGBC能誘導 AKT的磷酸化,起到信號傳導的作用(Fig 7B)。
為確定LncRNA-HGBC對AKT的誘導是否通過調控miR-502-3p實現的,在GSC-SD和EH-GB1細胞系中,作者通過突變LncRNA-HGBC上與 miR-502-3p結合的位點,發現過表達LncRNA-HGBC能誘導 AKT的磷酸化,以及N-鈣粘蛋白、波形蛋白的表達,進行突變的LncRNA-HGBC不能再對蛋白進行誘導(Fig 7C)。在EH-GB1細胞系中,將miR-502-3p導入過表達LncRNA-HGBC中會降低了這些蛋白的表達(Fig 7D);MK2206作為AKT抑制劑能逆轉LncRNA-HGBC對N-鈣粘蛋白和波形蛋白表達的影響(Fig 7E),表明LncRNA HGBC需要AKT激活。
為了進一步分析LncRNA-HGBC-miR-502-3p-SET-AKT之間的關聯,作者在43對GBC組織中進行分析,發現LncRNA HGBC與miR-502-3p呈負相關,與SET和HuR mRNA水平呈正相關(Fig 7F),與非腫瘤組織相比,HuR在GBC組織中表達上調,通過IHC分析發現,與非腫瘤組織相比,SET或p-AKT在GBC組織中表達上調(Fig 7G),此外,LncRNA -HGBC表達與SET和p-AKT表達呈正相關(Fig 7H and G)。綜上所述,LncRNA HGBC能通過競爭性結合miR-502-3p發揮作用,然后激活下游SET和AKT表達,從而加強腫瘤細胞的侵略性。
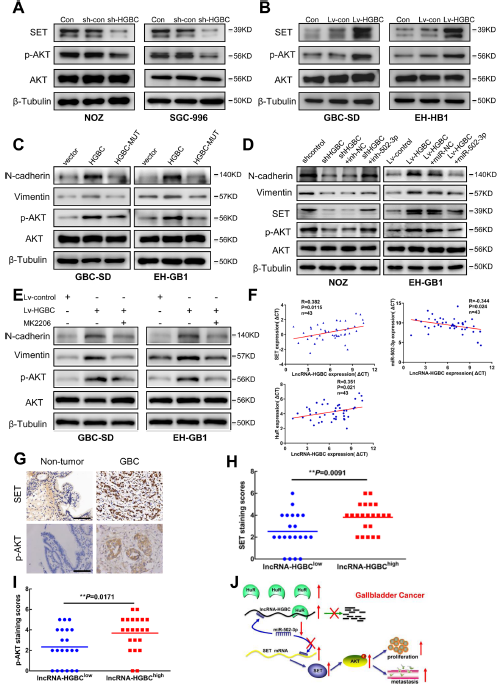
Figure 7 AKT是SET的下游效應物、lncRNA與miR-502-3p、SET和p-AKT的關系
本文發現了一個與GBC預后差相關的LncRNA-HGBC,并闡明了LncRNA-HGBC-miR-502-3p-SET-AKT通路介導GBC增殖、遷移、侵襲的分子機制。
參考文獻:
Hu Y P , Jin Y P , Wu X S , et al. LncRNA-HGBC stabilized by HuR promotes gallbladder cancer progression by regulating miR-502-3p/SET/AKT axis[J]. Molecular Cancer, 2019, 18(1). DOI:10.1186/s12943-019-1097-9.