






在HER2陽性乳腺癌中,PTEN和ATM軸控制G1/S細胞周期檢查點和腫瘤發生

PTEN包含一個n端磷酸酶結構域,它可以使脂質細胞膜的一種成分——磷脂酰肌醇3,4,5-三磷酸(PI(3,4,5)P3或PIP3)去磷酸化。PTEN通過去磷酸化PIP3的D3位,拮抗磷脂酰肌苷3-激酶(PI3K)通路。PI3K通路調節多種細胞過程,包括細胞代謝、存活、增殖、凋亡、生長和遷移。這些基本的細胞過程,當解除控制時,可以促進或驅動惡性表型。
PTEN功能突變的體細胞丟失在多種人類癌癥中被發現,包括乳腺癌、子宮內膜癌、多形性膠質母細胞瘤、皮膚癌和前列腺癌。PTEN缺失是乳腺癌中常見的事件,與加速進展和不良預后密切相關。特別是PTEN的表達已經被提出在人表皮生長因子受體2 (HER2)過表達乳腺癌中發揮重要作用。HER2是表皮生長因子受體家族的一員,具有酪氨酸激酶活性。它的過表達,在大約15 - 20%的乳腺癌病例中觀察到,與侵襲性臨床行為和不良預后相關。曲妥珠單抗是一種與HER2胞外結構域具有高親和力的單克隆抗體,是一種有效的治療HER2陽性乳腺癌患者的方法。PTEN表達檢測的總有效率約為70%,而PTEN表達陰性患者的總有效率僅為20%。
PTEN功能的喪失通常與基因組的不穩定性有關。此外,小鼠胚胎成纖維細胞(mef)中PTEN基因的缺失導致了未修復的DNA雙鏈斷裂的積累。PTEN缺失被認為通過至少兩種分子機制促進了基因組的完整性。在細胞核中,PTEN與著絲粒結合蛋白CENP-C結合,促進著絲粒組裝和中期到后期的轉變。此外,作為轉錄因子E2F1的輔助因子,核PTEN似乎調節Rad51的表達,Rad51是DNA修復機制的關鍵組成部分。然而,后續的研究得出了不一致的結果,表明PTEN在轉錄水平上對RAD51的調控可能僅限于特定的細胞環境。
PTEN缺陷改變了多個細胞周期檢查點,可能留下更少的時間進行DNA損傷修復和/或染色體分離。細胞周期的進展需要幾個分子過程的完美執行,以確保一個熟練的,無錯誤的,細胞分裂。這些事件發生的速度由周期蛋白依賴激酶(CDKs)的活性決定,CDKs磷酸化關鍵底物以促進DNA合成和有絲分裂進程。CDKs的催化活性受細胞周期檢查點的調控,這些檢查點監測細胞周期中主要事件的有序執行。檢查點代表了故障安全機制,它確保只有在滿足的最佳情況下才允許細胞分裂。所有生物體都需要通過細胞分裂周期進行適當的基因組維護,以確保正常生殖、發育和預防包括癌癥在內的各種疾病。DNA損傷可由內源性過程引起,如DNA復制過程中偶爾引入的DNA不匹配,拓撲異構酶I和拓撲異構酶II活性失效導致的DNA鏈斷裂,或由正常代謝副產品產生的ROS攻擊DNA。外源性來源主要包括誘變化學品、紫外線和電離輻射(IR)。細胞周期檢查點能夠檢測DNA損傷,提示其存在,并激活延緩細胞周期進程的通路,修復DNA損傷,或通過誘導細胞死亡來消除基因不穩定細胞。
哺乳動物細胞DNA損傷反應(DDR)信號的核心是共濟失調毛細血管擴張癥突變(ATM)和ATM-和rad3相關(ATR)蛋白激酶。ATM和ATR磷酸化并激活另外兩個激酶CHK1和CHK2, CHK1和CHK2與ATM和ATR一起,是細胞周期檢查點[24]的主調節器。通過調節CDKs的活性,這些分子在細胞周期G1 S期、S內期和G2 M期減緩或阻止細胞周期進程,使DNA損傷得以修復。ATM和ATR通過控制不同因子在DNA損傷位點的表達、活性或招募來促進DNA修復。一般來說,DDR機制能夠修復細胞在其生命周期中積累的DNA損傷,但如果認為損傷程度過高,就會誘導細胞凋亡或細胞衰老導致細胞死亡。
2021年4月,加拿大University Ave和中國香港大學團隊與癌癥研究所表觀遺傳學和基因組穩定性小組在Cell Death & Differentiation 雜志上發表了文章“The PTEN and ATM axis controls the G1/S cell cycle checkpoint and tumorigenesis in HER2-positive breast cancer”。此報道發現ATM磷酸化PTEN的398位(人類蘇氨酸;在小鼠絲氨酸)的激活,為了理解ATM磷酸化PTEN的生物學意義,建立了一個小鼠模型,構建了一個不能被ATM磷酸化的PTEN突變形式,用398位(PTEN- 398a)的絲氨酸取代了丙氨酸。PTEN-398A的表達可加速her2陽性乳腺癌小鼠模型的腫瘤發展和進展。利用分子生物學方法,發現了一種新的機制,通過ATM磷酸化PTEN調節其細胞再分配,并有助于該蛋白的腫瘤抑制功能。
技術路線:

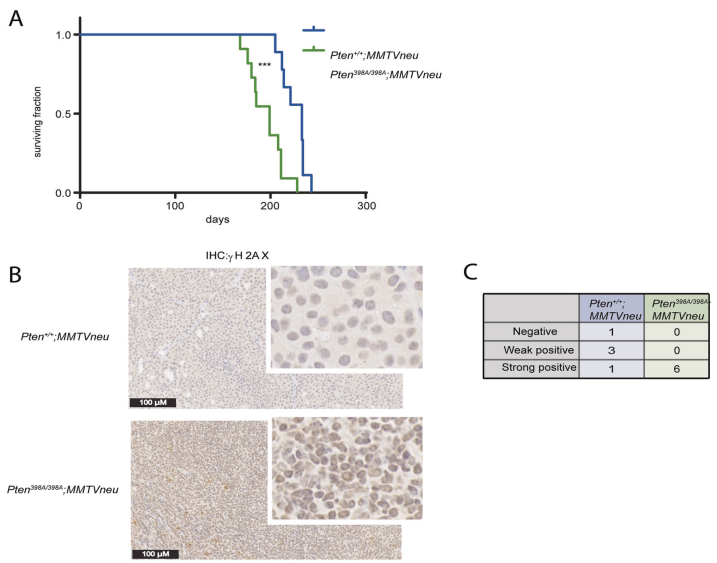
為了研究ATM磷酸化PTEN的體內功能,我們在小鼠中設計了一個敲入等位基因,其中398位絲氨酸取代了丙氨酸(Pten398A)。Pten398A/+和Pten398A/398A小鼠存活,發育明顯正常。為了研究atm依賴的PTEN磷酸化在乳腺癌中的可能作用,我們在小鼠乳腺腫瘤病毒(MMTV)啟動子/增強子轉錄控制下表達滅活neu (Erbb2)融合基因的小鼠背景下培養了Pten398A等位基因。在這個成熟的模型中,MMTVneu基因在正常乳腺上皮中低水平表達,導致乳腺腫瘤的發展,據報道中位發生率為205天。Pten+/+;MMTVneu小鼠發生了預期潛伏期的乳腺腫瘤,顯示了233天的中位生存期(圖1A)。相比之下,Pten398A/398A;MMTVneu小鼠腫瘤發展加快,中位生存期縮短至199天(圖1A)。對Pten+/+;MMTVneu和Pten398A/398A;MMTVneu小鼠的石蠟包埋腫瘤標本進行免疫組化檢測γH2AX。采用半定量方法對γH2AX免疫反應性進行評分(表1)。Pten+/+;MMTVneu腫瘤一般很少表達γH2AX(圖1B)。所有Pten398A/398A;MMTVneu樣本均為γH2AX強陽性,表明更高水平的基因組不穩定性(圖1B, C)。我們通過對腫瘤樣本的western blot分析證實了這些結果,結果顯示Pten398A/398A;MMTVneu腫瘤比Pten+/+;MMTVneu腫瘤表達更高水平的γH2AX(圖1D, E)。通過對Ki-67進行免疫組化來評估腫瘤的增殖指數,Ki-67是常規病理中廣泛使用的標志物。兩種基因型Pten+/+腫瘤的Ki67免疫反應性無顯著差異;MMTVneu(補充圖2C, D)。總之,Pten398A突變加速mmtv神經驅動的腫瘤發生,這與更高的基因組不穩定性有關,但在細胞增殖中沒有明顯的改變。
二、阻斷atm依賴的PTEN磷酸化導致基因組

為了確定ATM介導的PTEN磷酸化如何調節DDR,構建Pten398A/398A和Pten+/+ littermates MEFs細胞模型。PTEN蛋白水平不受398A突變的影響(補充圖3A, B)。此外,磷酸化AKT (PI3K通路活性的標記物)水平在Pten398A/398A和PTEN +/+ MEFs中相似(補充圖3B)。構建人乳腺上皮細胞系(MCF10A),穩定表達野生型PTEN (PTEN- wt),或PTEN的突變形式,不能被ATM磷酸化(PTEN- 398a)。在這些細胞中,內源性的PTEN基因被CRISPR/Cas9編輯刪除,創建了PTEN空細胞,然后用野生型或突變型的蛋白質穩定轉導重組。在表達PTEN- wt和PTEN- 398a的MCF10A細胞中,PTEN蛋白和磷酸化AKT水平相似(補充圖3A),使這些細胞成為ATM磷酸化PTEN分子作用研究的合適替代模型。
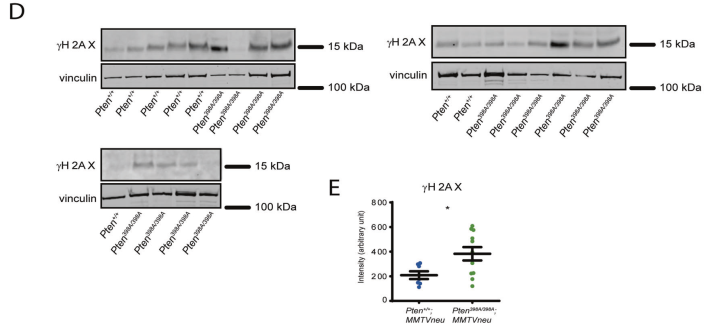
Pten+/+和Pten398A/398A mef在10gy IR作用下,通過測定每個細胞的γH2AX和53bp1病灶數量來評估其修復DNA損傷的能力。在Pten+/+ mef中,我們觀察到在照射后4小時,每個細胞的病灶數量達到峰值,24小時后恢復到接近基線水平(圖2A C)。Pten+/+和Pten398A/398A在ir后4小時,每個細胞聚集的病灶數量相似。然而,Pten398A/398A mef在24小時時間點比Pten+/+ mef保留了更多的病灶數,表明DNA修復能力降低(圖2 C)。
三、pten - s38a突變誘導細胞凋亡抵抗

為了進一步了解阻斷atm依賴的PTEN磷酸化如何影響細胞對基因毒性損傷的反應,我們使用抗體陣列評估了表達PTEN- wt或PTEN- 398a的MCF10A細胞裂解液的應激反應和凋亡通路的活性。Pten+/+和Pten398A/398A MEF培養,表達Pten - wt的MCF10A細胞與表達Pten -398A的MCF10A細胞在正常培養條件下的生長速率均無差異(圖3A和補充圖5C)。對細胞進行不同的基因毒性脅迫處理:IR、順鉑和柔紅霉素。在所有條件下,Pten398A/398A MEFs和PTEN-398A表達的MCF10A細胞對基因毒性脅迫的抗性均高于野生型PTEN的細胞(圖3B D和Supplementary Fig. 5D, E)。通過流式細胞儀Annexin V染色判斷(圖3E)。通過使用caspase-3抗體對Pten+/+、MMTVneu和Pten398A/398A、MMTVneu腫瘤進行免疫組化染色,證實了這些觀察結果。caspase-3抗體是一種廣泛用于檢測凋亡細胞的標志物。確實,MMTVneu腫瘤的caspase-3陽性細胞比相應的Pten+/+少;綜上所述,這些結果表明,PTEN不能被ATM磷酸化的細胞對基因毒性應激誘導的細胞凋亡具有抗性。為了評估表達PTEN-398A且DNA損傷未解決的細胞在輻照(圖2B, C和補充圖4B, C)后的持續存在是否是誘導凋亡反應失敗的結果,我們在IR之前先用泛caspase抑制劑z-VAD-FMK (zVAD)對細胞進行預處理。與DMSO處理相比,zVAD增加了PTEN-WT細胞中未解決的DNA損傷灶的數量,其水平與DMSO處理的PTEN-398A細胞相當(補充圖5F)。然而,需要注意的是,zVAD預處理也略微增加了IR后PTEN-398A細胞中未解決的DNA損傷灶的數量。
四、atm依賴性PTEN磷酸化缺失的細胞中G1/S細胞周期檢查點受損
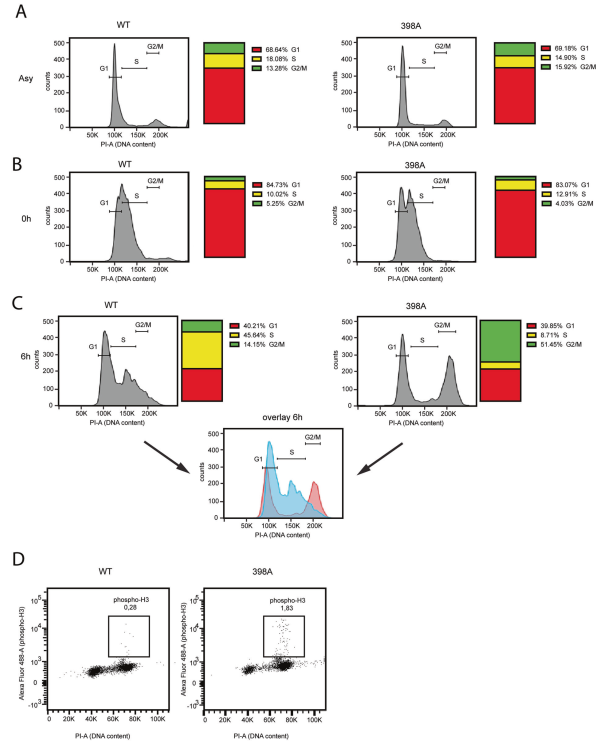
對表達PTEN-WT或PTEN-398A的MCF10A細胞進行了cDNA微陣列分析,并用順鉑誘導基因毒性應激。差異表達基因列表采用基因集合富集分析[39]進行分析。有趣的是,表達PTEN- 398A的細胞中上調的基因與G1/S檢查點的激活有關,如G1/S期特異性轉錄、E2F靶標、Rb1通路控制的細胞周期調控基因等推測表達PTEN-398A的細胞對基因毒性脅迫的抗性可能是由于未能阻止細胞周期以應對DNA損傷。為了測試這種可能性,測量了表達野生型或突變PTEN的同步細胞在細胞周期阻滯在G1/ S檢查點的能力,以應對DNA損傷。在表達PTEN-WT或PTEN-398A的正常周期細胞中,G1、S、G2/M期細胞比例沒有明顯差異(圖4A)。為了使細胞在G1/S檢查點同步,我們對它們施加雙胸腺嘧啶脈沖。這種處理有效地抑制了PTEN-WT和PTEN-398A細胞在G1/S邊界(圖4B)。然后在正常生長培養基中釋放細胞,在S期進展期間用IR處理,6小時后收集細胞,用流式細胞術分析細胞周期分布(實驗方案見補充圖7A)。大部分PTEN-WT細胞被阻滯在s期,而PTEN-398A細胞未能阻滯細胞周期,而是發展到G2/M期(圖4C)。通過測定表達磷酸化組蛋白H3 (Ser10)的細胞比例,證實了這一觀察結果。磷酸化組蛋白H3 (Ser10)標志著細胞處于有絲分裂階段。(圖4 d)。
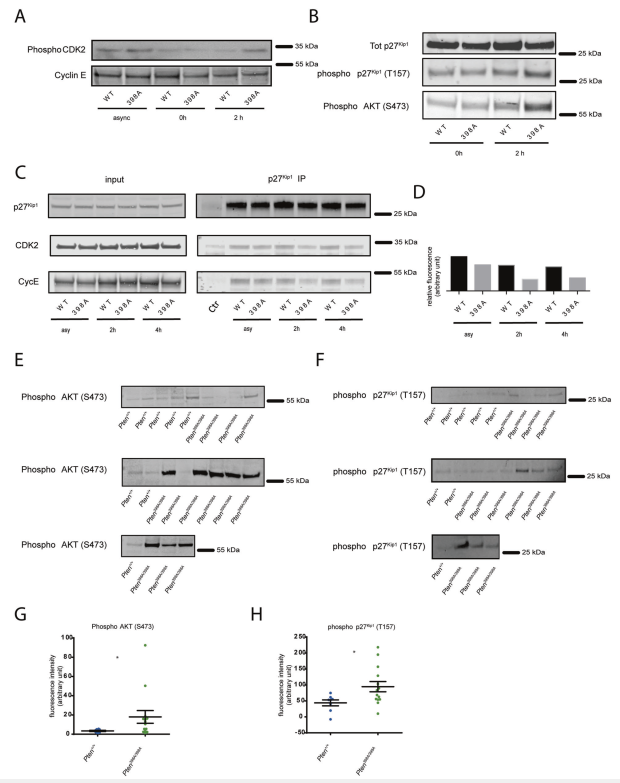
表達PTENWT的細胞,在雙胸苷激酶阻斷和輻射釋放后被阻滯在S期,表現出低水平的CDK2磷酸化(圖5A)。相反,經過相同處理的PTEN-398A細胞有更高水平的磷酸化CDK2(圖5A),這與DNA損傷檢查點的缺陷激活一致。與PTEN-WT細胞相比,PTEN-398細胞中磷酸化的AKT和磷酸化的p27Kip1水平更高(圖5B)。雖然p27Kip1與CDK2和Cyclin E在照射過的PTEN-WT細胞中有效地共免疫沉淀,但在PTEN-398A細胞中這些相互作用減弱(圖5C, D)。與我們在MCF10A細胞中觀察到的結果一致,免疫印跡分析顯示PTEN398A/398A裂解物中磷酸化的AKT和磷酸化的p27Kip1水平更高;MMTVNeu腫瘤與Pten+/+;MMTVNeu對應腫瘤進行比較(圖5E-H)。總之,抑制ATM對PTEN的磷酸化增加了DNA損傷后AKT和p27Kip1的激活,反過來有利于CDK2/Cyclin E復合物的活性,并驅動細胞周期進程。
五、阻斷atm依賴的磷酸化會損害DNA損傷后PTEN的亞細胞再分配
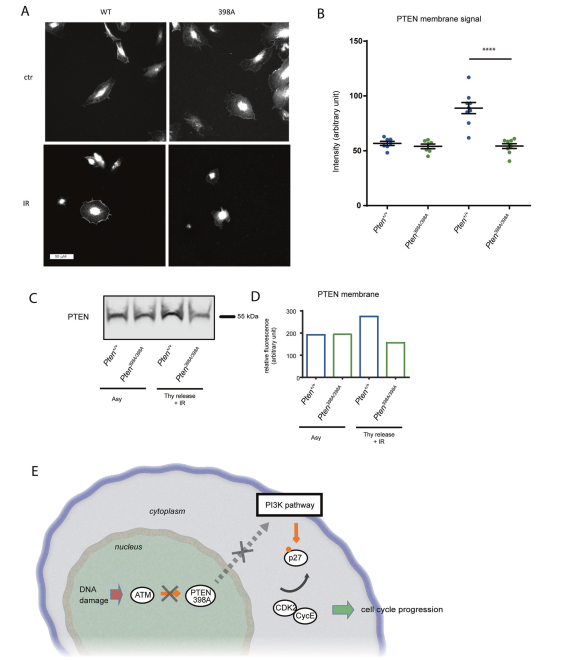
研究了磷脂酰肌醇3,4,5磷酸(PIP3)在DNA損傷后的細胞定位。將MCF10A細胞按上述方法同步輻照,免疫熒光法分析PTEN的亞細胞分布。PTEN-WT和PTEN-398A蛋白在細胞核和質膜穩定定位(圖6A, B)。經過IR處理后,PTEN-WT在質膜上積累(圖6A, B)。相比之下,在這些條件下,沒有檢測到PTEN-398A蛋白的重新定位(圖6A, B)。B).我們通過細胞分離和膜相關PTEN的免疫印跡分析進一步證實了這些結果(圖6C, D)。這些結果表明,ATM對PTEN的磷酸化導致PTEN在質膜上重新分布,這與AKT通路激活減少有關。