






YTHDF1通過以m6A依賴的方式促進肝細胞癌的進展
肝細胞癌(HCC)是最具侵襲性的惡性腫瘤之一,是全球第四大癌癥相關死亡原因。新的證據表明,m6A在腫瘤進展中起著關鍵作用。然而,YTHDF1在HCC的生物學功能仍不清楚。發表于影響因子7.032的“Mol Ther Nucleic Acids”的“YTHDF1 Facilitates the Progression of Hepatocellular Carcinoma by Promoting FZD5 mRNA Translation in an m6A-Dependent Manner”詳細介紹了YTHDF1在HCC中的生物學功能及作用機制。在這里,我們發現在HCC組織和細胞系中YTHDF1的表達顯著升高,并且與HCC患者的預后顯著相關。此外,在HCC中,YTHDF1的表達受USF1和c-MYC轉錄調控。功能研究表明,YTHDF1在體內外均可促進HCC細胞的增殖和轉移。多組學分析顯示,YTHDF1可以以m6A依賴的方式加速FZD5 mRNA的翻譯輸出,并通過WNT/β-catenin途徑作為癌基因發揮作用。

技術路線:
結果:
(1) YTHDF1表達在HCC中顯著上調并與不良預后相關
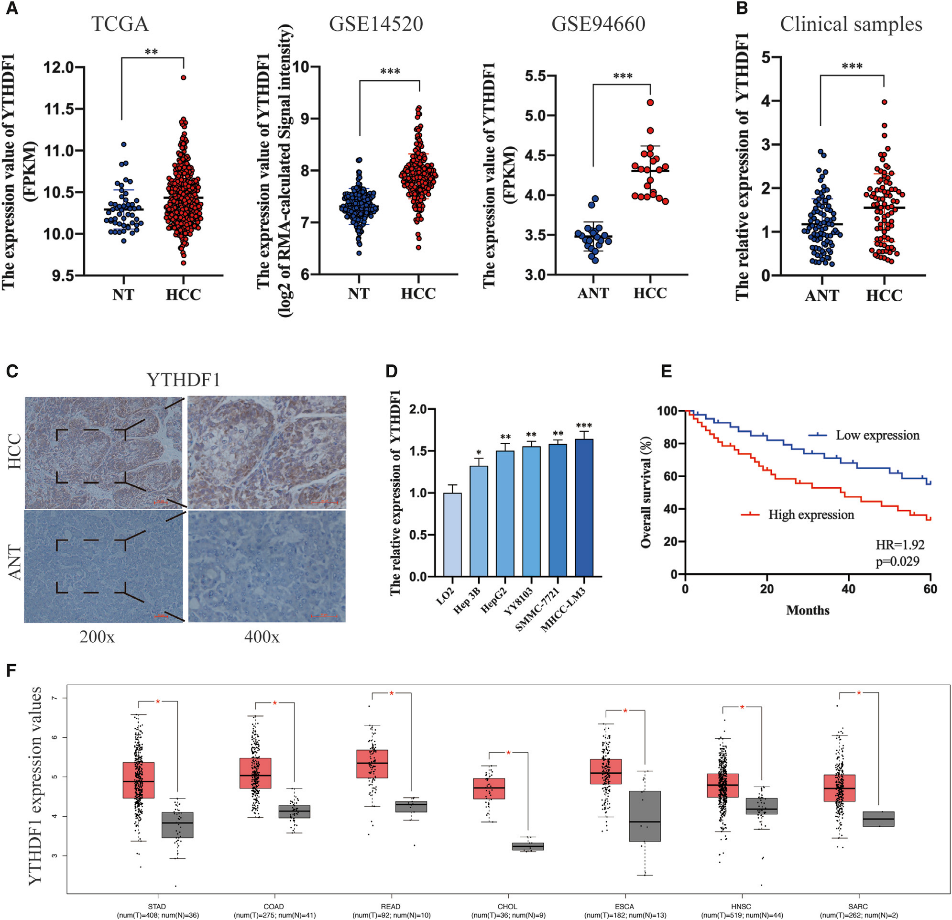
為了探索YTHDF1在HCC的調節作用,我們首先分析了它的表達。TCGA數據庫、GSE14520數據集和GSE94660數據集顯示YTHDF1在HCC組織中的表達顯著上調(圖1A)。此外,我們通過臨床樣本的qRT-PCR和免疫組織化學(IHC)分析檢測了YTHDF1的表達,與相鄰正常組織相比,HCC組織中YTHDF1在mRNA和蛋白水平上均過表達(圖1B和1C)。此外,qRT-PCR分析證實,與LO2細胞相比,HCC細胞中的YTHDF1表達升高(圖1D)。為探討YTHDF1表達與臨床病理特征的相關性,我們根據YTHDF1表達的中值將HCC患者分為兩組。用Kaplan-Meier法進行的生存分析表明,高YTHDF1表達的HCC患者經歷了更糟糕的OS(圖1E)。有趣的是,與相應的正常組織相比,在多種實體惡性腫瘤中發現YTHDF1的表達顯著上調(圖1F)。根據上述數據,我們得出結論,YTHDF1的表達在HCC顯著上調,并與HCC患者的不良預后相關。
(2) YTHDF1的表達由USF1和c-MYC共同控制
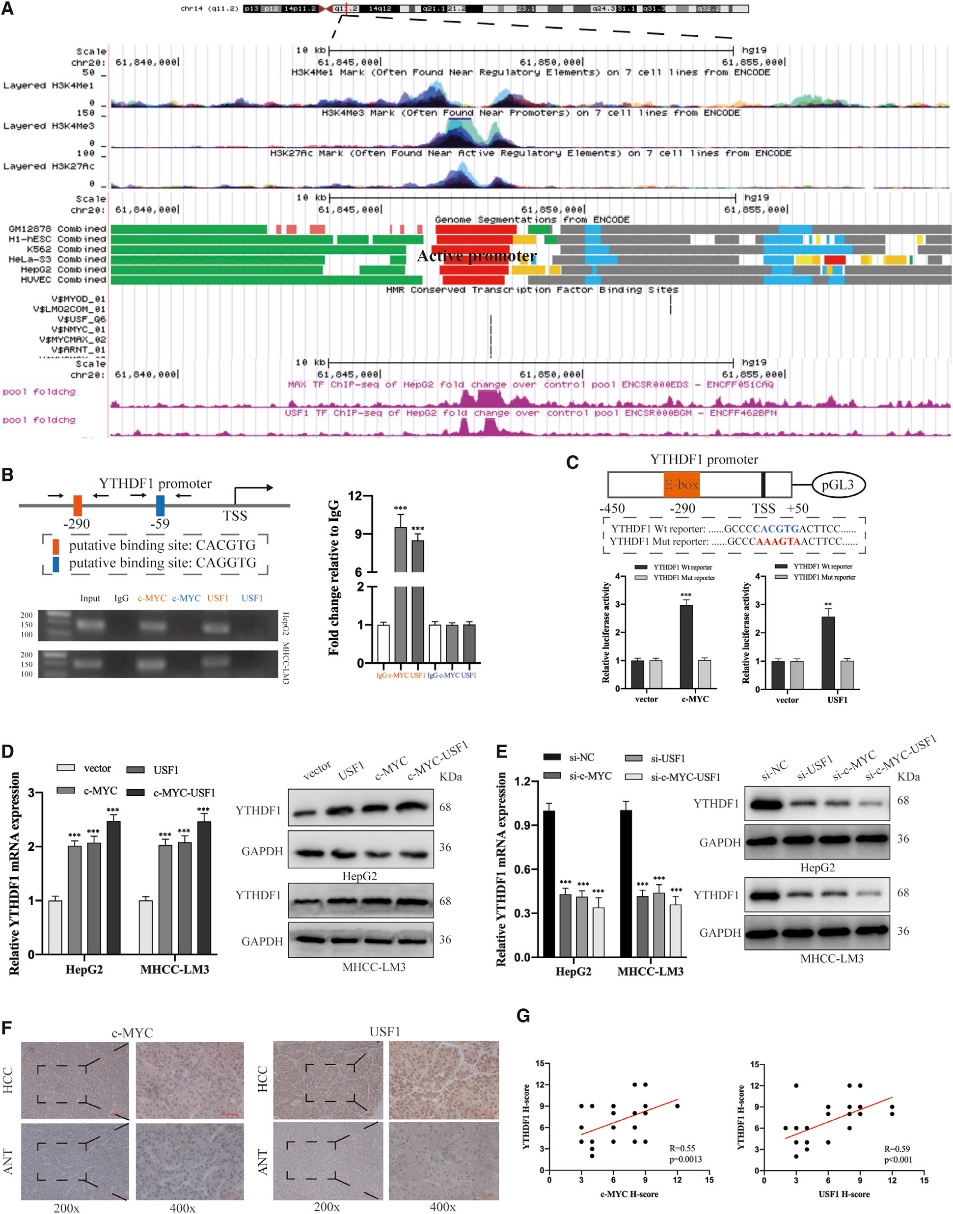
鑒于轉錄因子在基因表達中的廣泛調節作用,我們旨在研究在YTHDF1的表達是否受到特定轉錄因子的調節。UCSC基因組瀏覽器數據庫顯示YTHDF1啟動子在多種細胞系中非常活躍,包括HepG2細胞(圖2A)。我們接下來分析了從ENCODE數據庫下載的HepG2細胞ChIP-seq數據。如圖2A所示,在YTHDF1啟動子中c-MYC/MAX和USF1的潛在結合(圖2A)。c-MYC/MAX和USF1位于E-box的290bp (圖2B)。此外,熒光素酶活性顯示,在HEK293T細胞中用c-MYC或USF1過表達載體共轉染YTHDF1啟動子報告子后,只有野生型推定結合位點增加了YTHDF1啟動子的激活(圖2C)。qRT-PCR和免疫印跡分析表明,c-MYC/USF1的缺失減少了但過表達增加了YTHDF1在基因和蛋白質水平上的表達,當c-MYC和USF1都受到調節時,這種表達更為有效(圖2D和2E)。IHC分析顯示,與ANTs相比,c-MYC和USF1在HCC組織中的表達顯著上調,并與YTHDF1的表達呈正相關(圖2F和2G)。綜上所述,在HCC,YTHDF1的表達由USF1和c-MYC共同控制。
(3) YTHDF1促進HCC細胞體外增殖和轉移
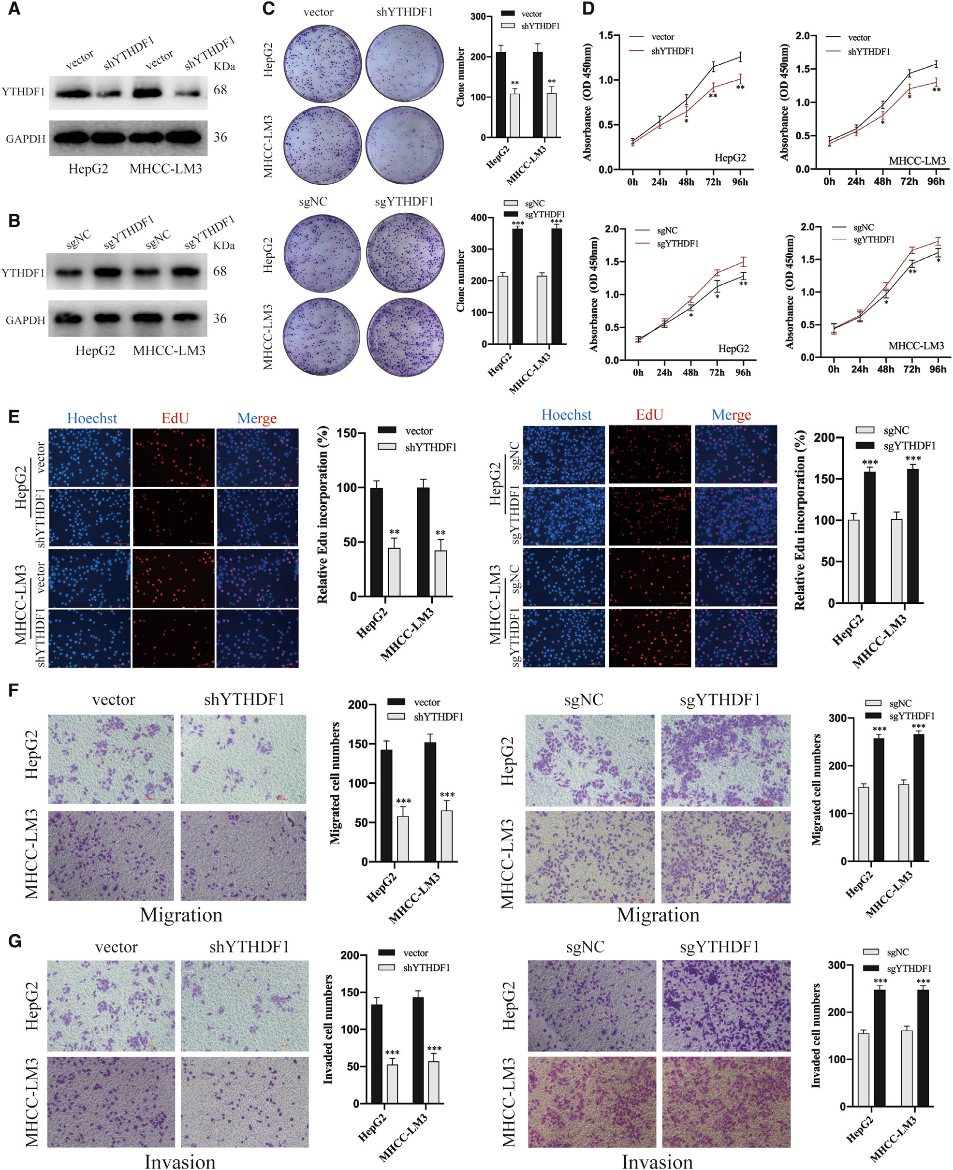
為了研究YTHDF1在HCC的生物學功能,我們設計了慢病毒介導的shYTHDF1和CRISPR-dCas9基因激活系統,分別在HCC細胞中敲除和過表達YTHDF1的表達(圖3A和3B)。集落形成試驗顯示,YTHDF1敲除明顯抑制,但YTHDF1過表達顯著增強HCC細胞的集落形成能力(圖3C)。CCK-8分析和EdU分析顯示沉默YTHDF1可以抑制肝癌細胞的增殖,而上調YTHDF1可以促進肝癌細胞的增殖(圖3D和3E)。Transwell顯示,YTHDF1下調和上調分別顯著抑制和提高了HCC細胞的遷移和侵入能力(圖3F和3G)。上述結果表明YTHDF1在HCC細胞中發揮致癌作用。
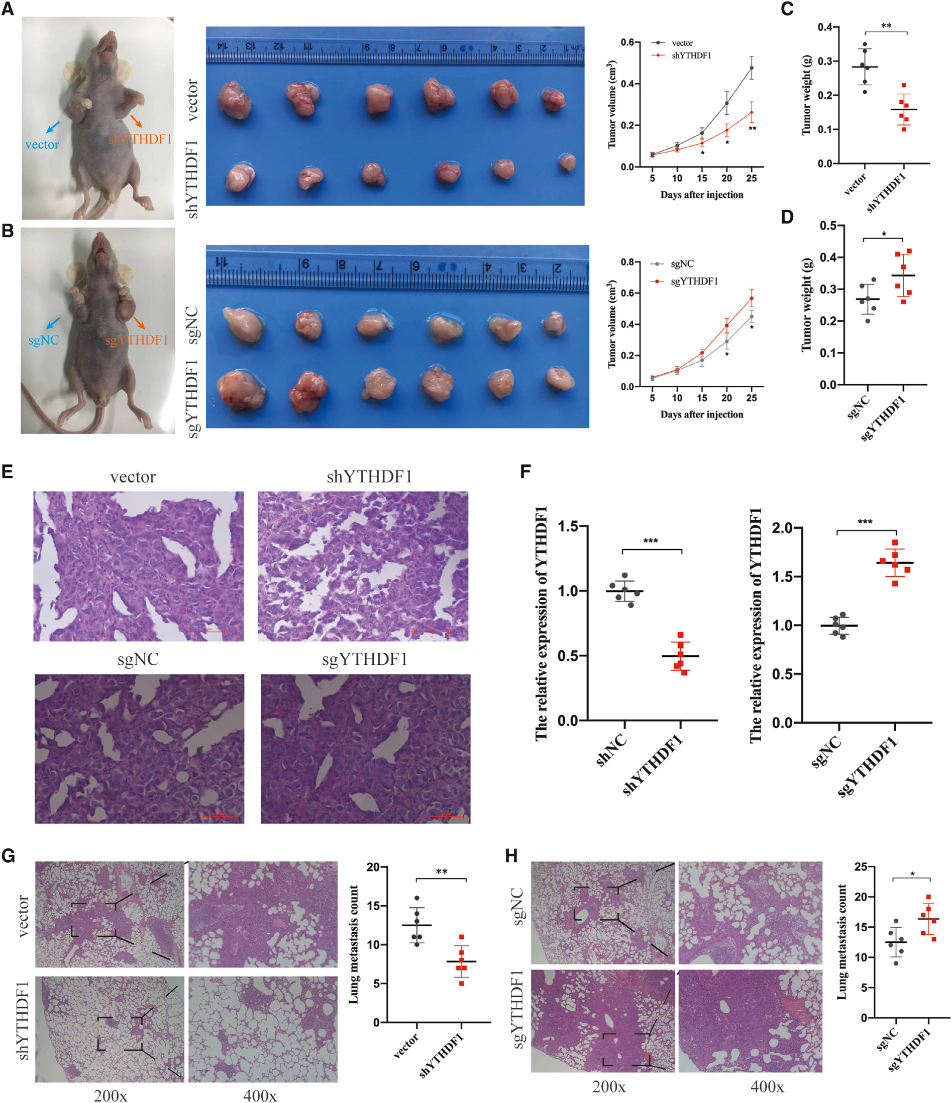
(4) YTHDF1促進體內HCC細胞增殖和轉移
為了研究YTHDF1在HCC體內的致癌作用,我們用YTHDF1沉默的MHCC-LM3細胞和YTHDF1過表達的HepG2細胞在裸鼠體內進行了皮下植入實驗。數據顯示,YTHDF1敲除有效減少,但YTHDF1過表達顯著增加腫瘤大小和重量(圖4A-4D)。并通過HE染色驗證各組腫瘤組織(圖4E)。接下來,進行qPCR分析以確認異種移植腫瘤組織中YTHDF1的表達。正如預期的那樣,YTHDF1沉默的MHCC-LM3細胞形成的腫瘤組織顯示出降低的YTHDF1表達,而來自YTHDF1過表達的HepG2細胞的腫瘤組織顯示出增加的YTHDF1表達(圖4F)。為了評價YTHDF1是否能促進體內轉移,我們通過靜脈注射YTHDF1沉默的MHCC-LM3細胞和YTHDF1過表達的HepG2細胞建立了體內轉移模型。我們觀察到,與對照組相比,YTHDF1敲除組肺表面轉移結節較少,而YTHDF1過表達組肺表面轉移結節較多(圖4G和4H)。總的來說,我們的發現揭示了YTHDF1促進體內HCC細胞的生長和轉移。
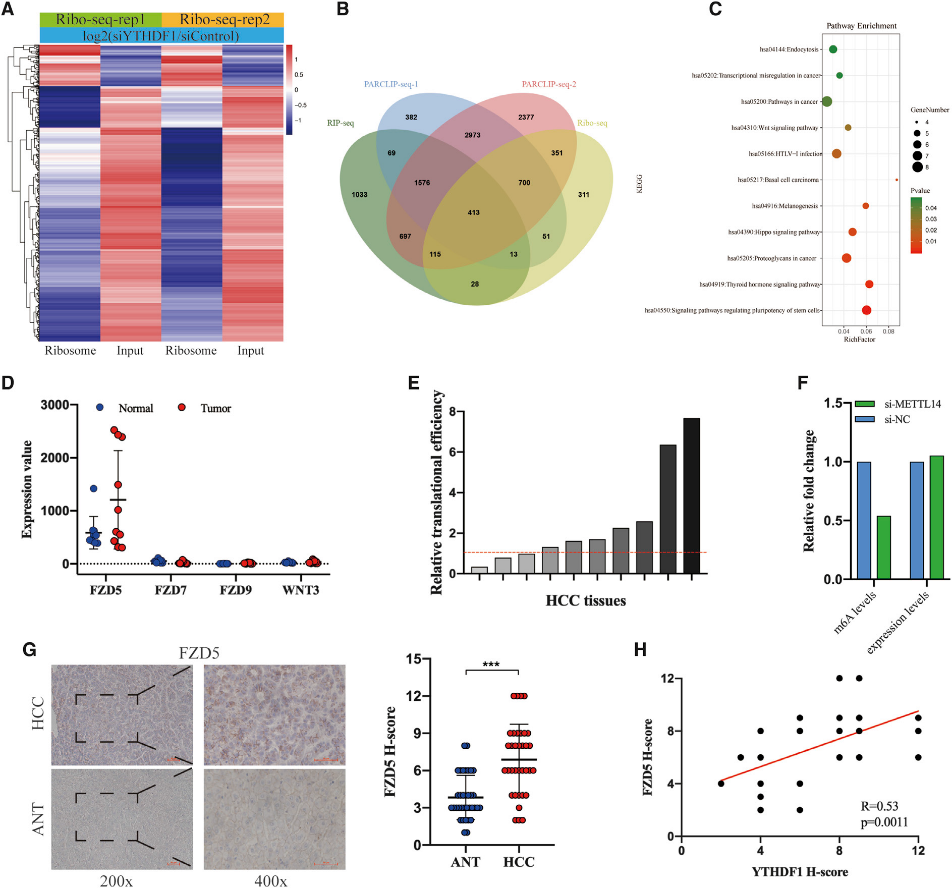
(5) 將FZD5確定為YTHDF1在HCC的直接目標
為了鑒定YTHDF1的靶標,我們首先分析了具有YTHDF1敲除的細胞的公共核糖體圖譜數據(GSE 63591)。YTHDF1的缺失導致1982個轉錄物的翻譯效率降低(圖5A)。為了選擇與YTHDF1直接結合的靶點,我們下載了GSE63591中YTHDF1的RNA RIP-seq和PARCLIP-seq數據,并與核糖體-seq數據重疊。我們發現YTHDF1的缺失導致與YTHDF1結合的413個基因的翻譯效率降低(圖5B)。KEGG分析表明,這些轉錄物在11種信號通路中顯著富集,如Hippo和WNT信號通路(圖5C),其中FZD5、FZD7、FZD9和WNT3在大多數通路中富集(8/11)。有趣的是,當我們使用GSE112705數據集在HCC中檢測FZD5、FZD7、FZD9和WNT3的表達豐度和翻譯效率時,我們發現FZD5的表達豐度比FZD7、FZD9和WNT3高得多,并且與ANTs相比,FZD5在大多數HCC組織中的轉錄效率得到提高(圖5D和5E)。考慮到m6A的修飾具有高度的細胞類型特異性,我們分析了GSE9064212中的HepG2細胞RNA-seq和m6A-seq數據,以驗證FZD5基因的m6A修飾,結果表明METTL14的敲除顯著降低了人肝癌細胞中m6A的水平,而不是FZD5基因的表達水平(圖5F)。IHC分析顯示,與ANTs相比,FZD5在HCC組織中的表達顯著上調(圖5G),并且與YTHDF1的表達呈強正相關(圖5H)。
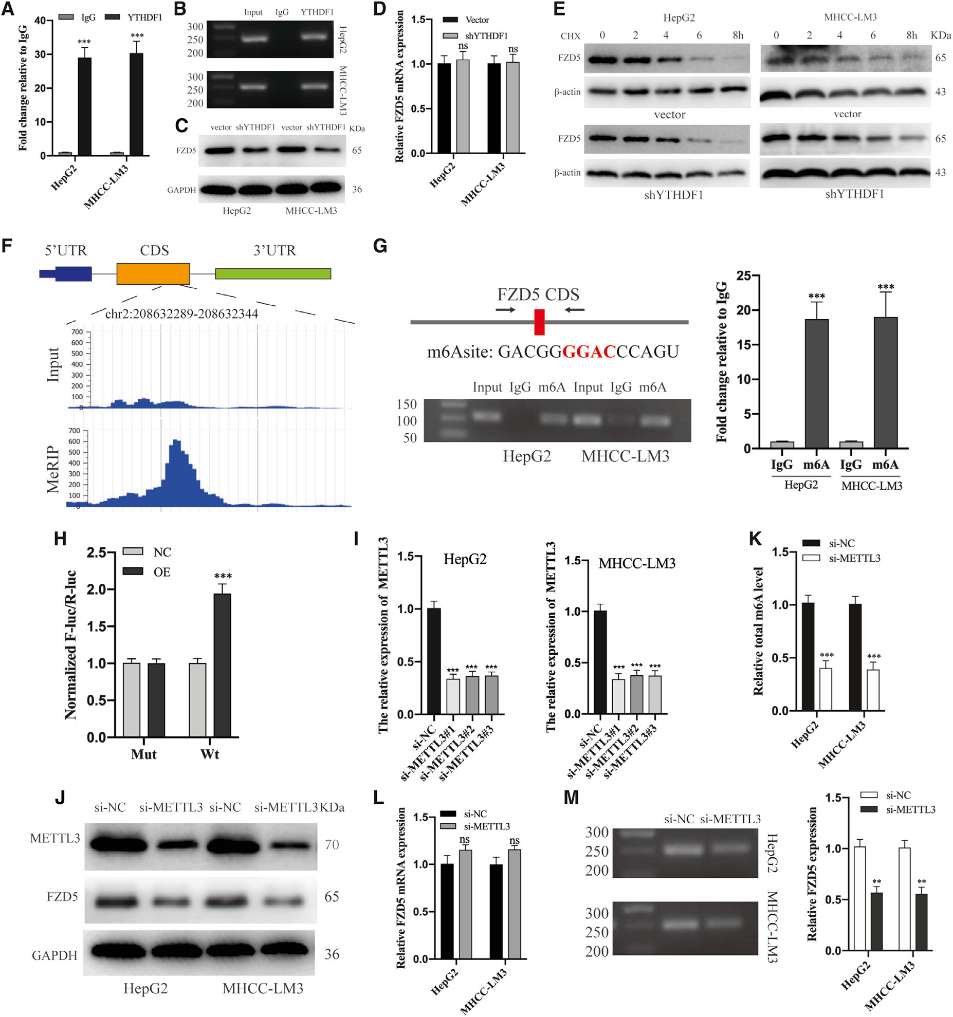
(6) YTHDF1以m6A依賴的方式促進FZD5 mRNA的翻譯
為了驗證FZD5基因和YTHDF1蛋白之間的直接結合,我們進行了RIP實驗。半定量PCR和qRT-PCR分析顯示與免疫球蛋白G相比,FZD5 mRNA與YTHDF1蛋白結合顯著富集(圖6A和6B)。此外,敲除YTHDF1導致FZD5在蛋白質水平上的顯著抑制(圖6C),但在mRNA水平上沒有(圖6D)。為了排除YTHDF1可能影響FZD5蛋白穩定性,我們用環己酰亞胺(CHX)處理HepG2和MHCC-LM3細胞以阻斷翻譯,并發現FZD5蛋白表達在載體組和shYTHDF1組中類似地被消除(圖6E)。上述結果表明,由YTHDF1沉默引起的FZD5蛋白表達的消除是由于mRNA翻譯效率的降低,而不是轉錄或蛋白穩定性的降低。
眾所周知,YTHDF1通過結合m6A甲基化轉錄物發揮作用。因此,我們首先使用MeT-DB V2.0數據庫篩選了HepG2細胞中FZD5基因的m6A位點,該數據庫顯示FZD5基因m6A位點富集在位于chr 2:208632313–208632314的蛋白質編碼序列(CDS)中(圖6F)。MeRIP-PCR證實了m6A修飾在該位點的顯著富集(圖6G)。此外,熒光素酶分析顯示,YTHDF1過表達增強了FZD5-WT的表達(圖6H)。此外,METTL3敲除(圖6I和6J)顯著降低了HCC細胞中m6A總水平(圖6K)和FZD5蛋白表達(圖6J),FZD5 mRNA表達略有上調(圖6L)。此外,RIP分析顯示,METTL3敲除顯著抑制了YTHDF1和FZD5 mRNA之間的結合(圖6M)。我們的結果表明,YTHDF1選擇性地識別FZD5基因CDS中的m6A位點,并隨后促進其翻譯輸出。
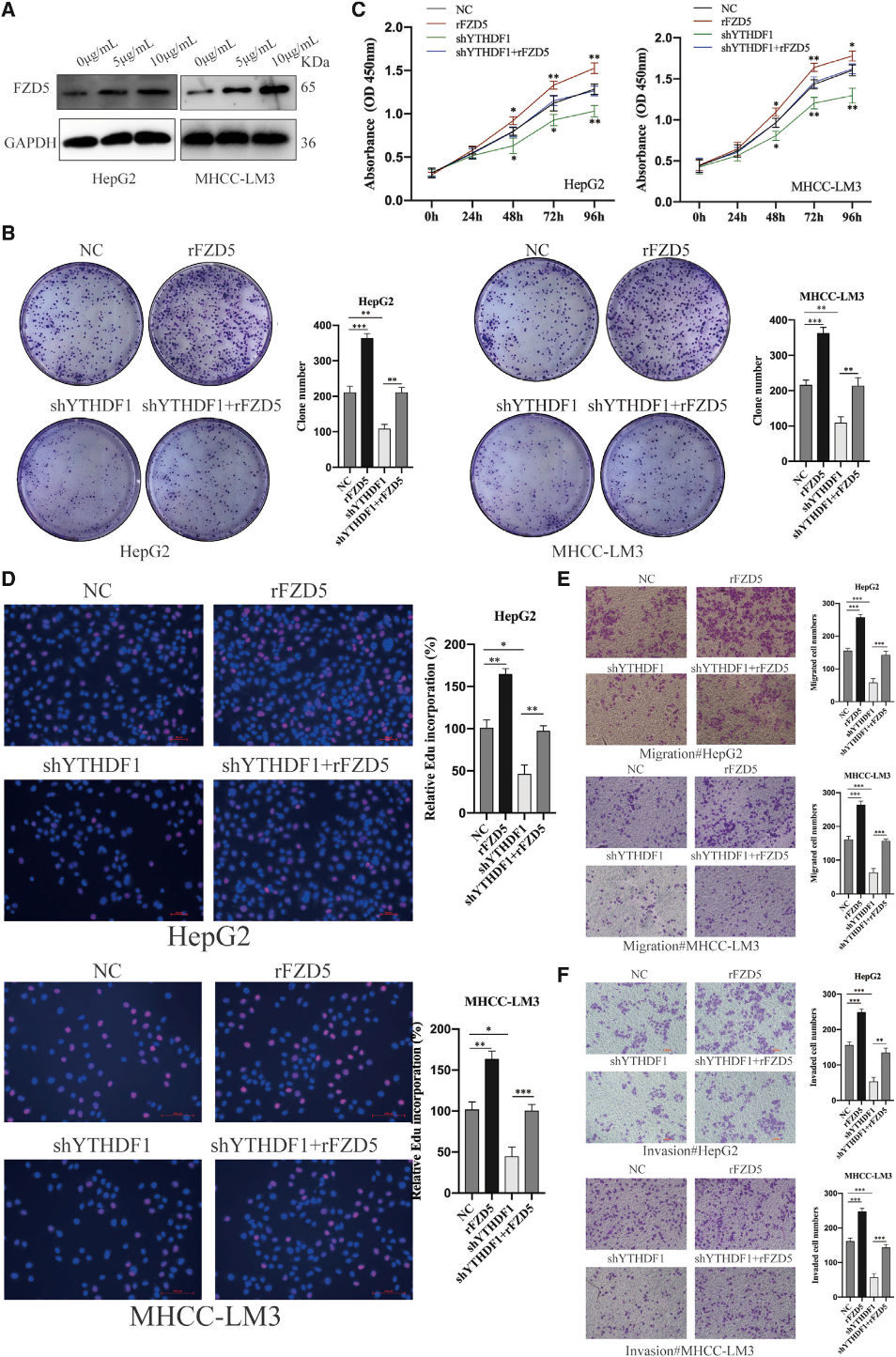
(7) FZD5過表達有效逆轉YTHDF1敲除誘導的HCC細胞進程抑制
為了探索FZD5在HCC的調節作用,我們通過轉染具有生物活性的重組人FZD5來過表達FZD5 (圖7A)。細胞表型顯示FZD5上調顯著促進了HCC細胞的增殖、遷移和侵襲(圖7B-7F)。接下來,我們在YTHDF1缺陷型HCC細胞中過表達FZD5,以研究YTHDF1的致癌作用是否由FZD5直接介導。集落形成試驗、CCK-8試驗和EdU試驗表明,YTHDF1敲除損害了細胞集落形成和生長,而FZD5上調逆轉了這種效應(圖7B-7D)。一致地,Transwell分析顯示,在HCC細胞中FZD5過表達后,由YTHDF1耗竭抑制的細胞遷移和侵入能力重新建立(圖7E和7F)。
(8) YTHDF1通過WNT/β-Catenin信號通路促進HCC癌的發生
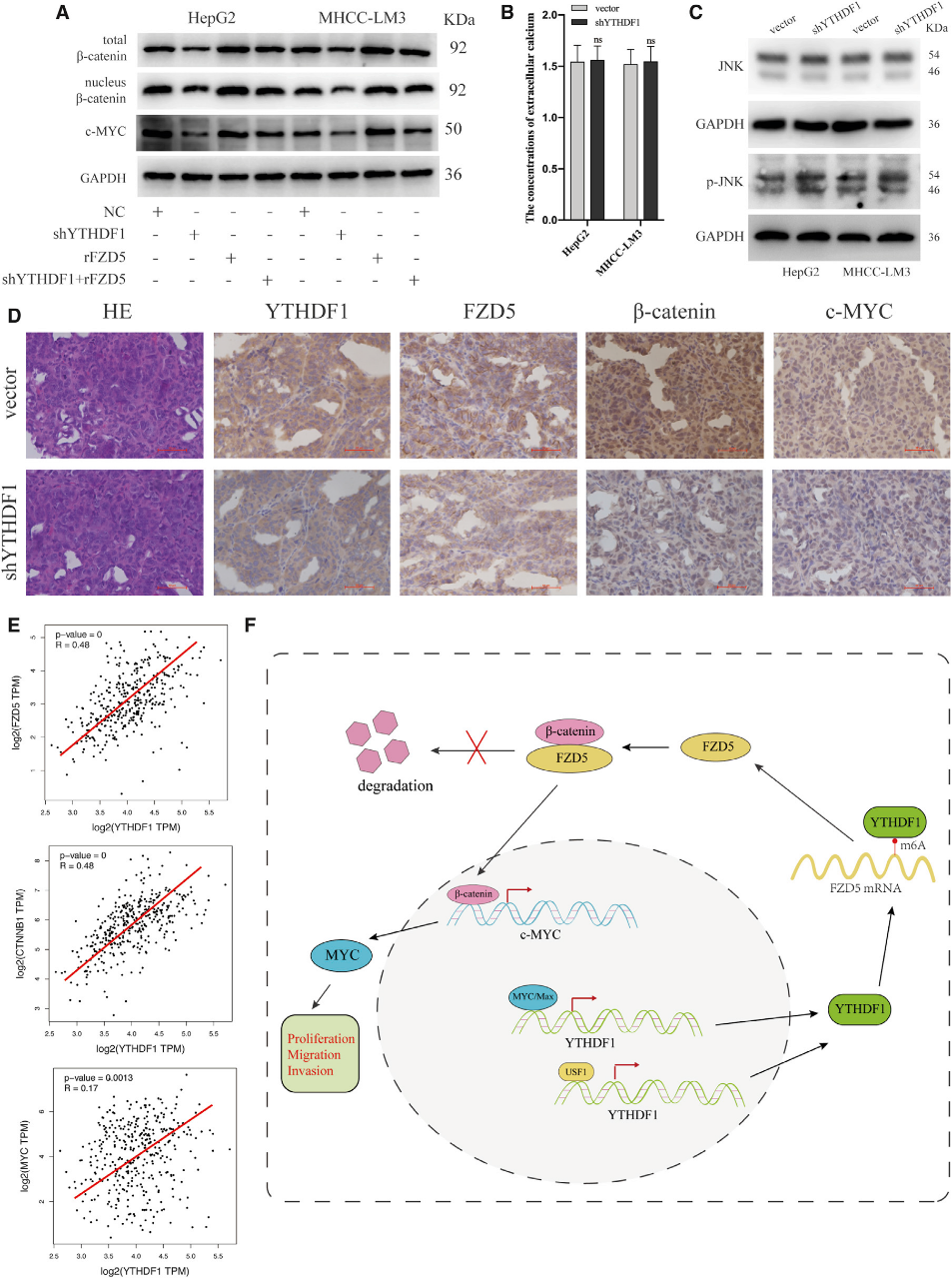
據報道,FZD5過表達在肝癌中激活WNT/β-Catenin途徑。接下來,我們研究了YTHDF1是否通過FZD5/WNT/β-Catenin發揮作用。亞細胞分級和免疫印跡分析顯示,YTHDF1的敲除顯著降低了HCC細胞中β-Catenin的總表達和核表達,而FZD5的上調逆轉了這種效應(圖8A)。此外,我們發現YTHDF1敲除不影響細胞外鈣的濃度(圖8B)和磷酸化JNK水平(圖8C)。這些結果證實了YTHDF1在HCC的致癌作用是通過WNT/β-Catenin途徑而不是WNT/Ca2+途徑或平面細胞極性途徑介導的。眾所周知,c-Myc基因是一種由WNT/β-Catenin信號轉錄激活的癌基因,它在很大程度上參與了各種癌癥的發展。免疫組化分析表明,YTHDF消耗抑制但FZD5的過表達重建了HCC細胞中c-MYC的表達(圖8A)。此外,IHC分析顯示,與陰性對照組相比,FZD5、β-Catenin和c-MYC在YTHDF1下調的異種移植腫瘤樣品中的表達顯著降低(圖8D)。此外,我們在HCC的TCGA數據庫中觀察到FZD5、β-Catenin、cMYC和YTHDF1表達呈正相關(圖8E)。綜上所述,我們的發現表明YTHDF1通過FZD5/WNT/β-Catenin信號通路促進HCC細胞的致癌作用(圖8F)。
結論:YTHDF1可以通過FZD5/WNT/β-Catenin信號通路,以m6A依賴的方式提高FZD5 mRNA的翻譯輸出,促進HCC細胞的進程。我們的研究為HCC提供了一個潛在的治療策略。