






如何從NCBI下載別人的測序原始數據
使用別人發表的高通量測序數據進行挖掘,篩選差異基因進行后續研究,不僅節省經費,又節約時間,可以大大提升研究人員的工作效率。今天小編就給大家介紹下如何從NCBI數據庫下載高通量測序數據。
1、進入NCBI SRA數據庫搜索項目界面,輸入SRP編號。SRP編號一般可以文章中找到。現在大部分雜志都需要作者上傳測序數據到公共數據庫,并將提交得到的編號SRP編號(測序)或GSE編號(芯片)放到文章中。
https://www.ncbi.nlm.nih.gov/Traces/study/

2、輸入SRP編號后出來界面會顯示測序數據信息,我們輸入SRP110184點擊搜索后,會找到8個樣本的測序數據。

3、數據下載,第一可以直接點擊單個樣本,進行頁面下載。Data access界面有兩個下載鏈接都可以下載。
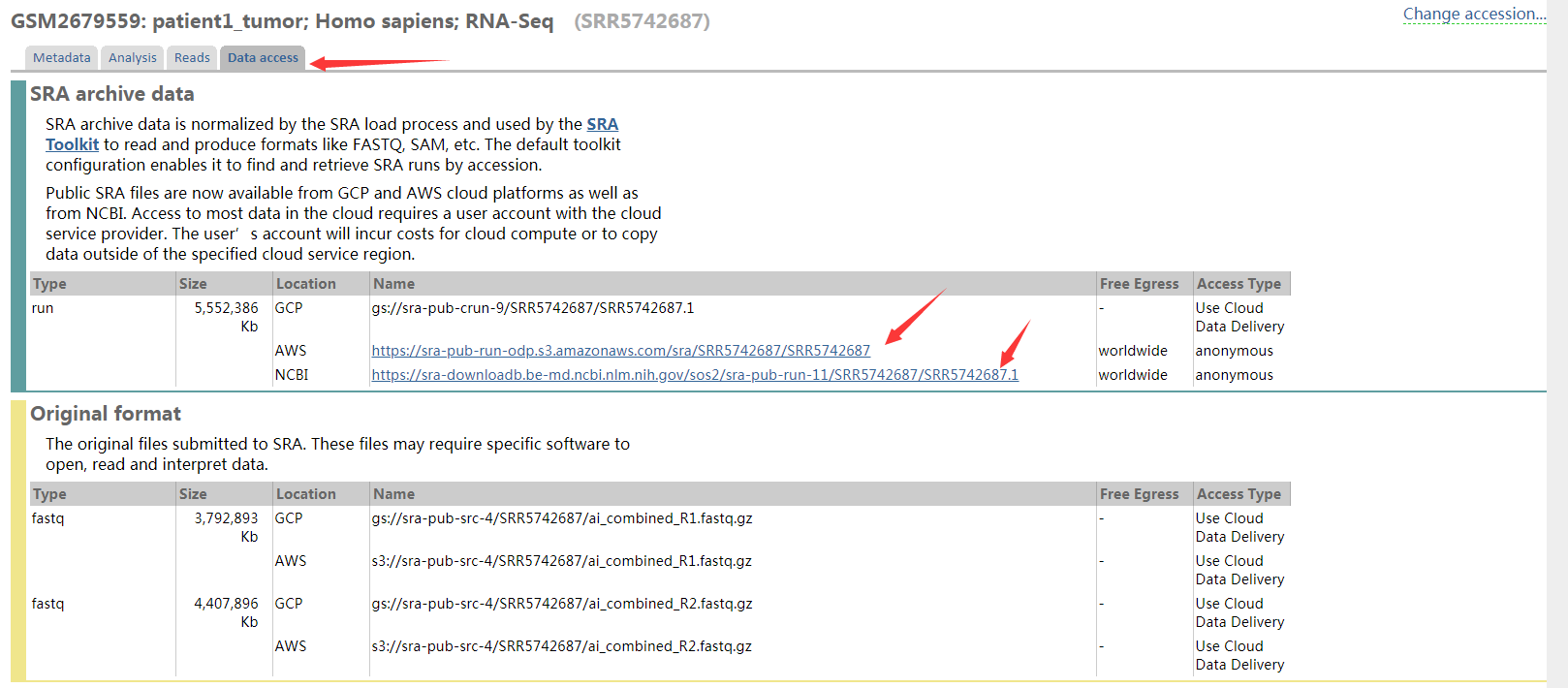
4、下載的數據需要轉化為fastq格式才能進行下一步的差異表達分析。這里就需要NCBI提供的下載工具SRA Toolkit進行轉化。AWS,NCBI兩個鏈接下載的文件后綴為”.man”,”.1”格式。
https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
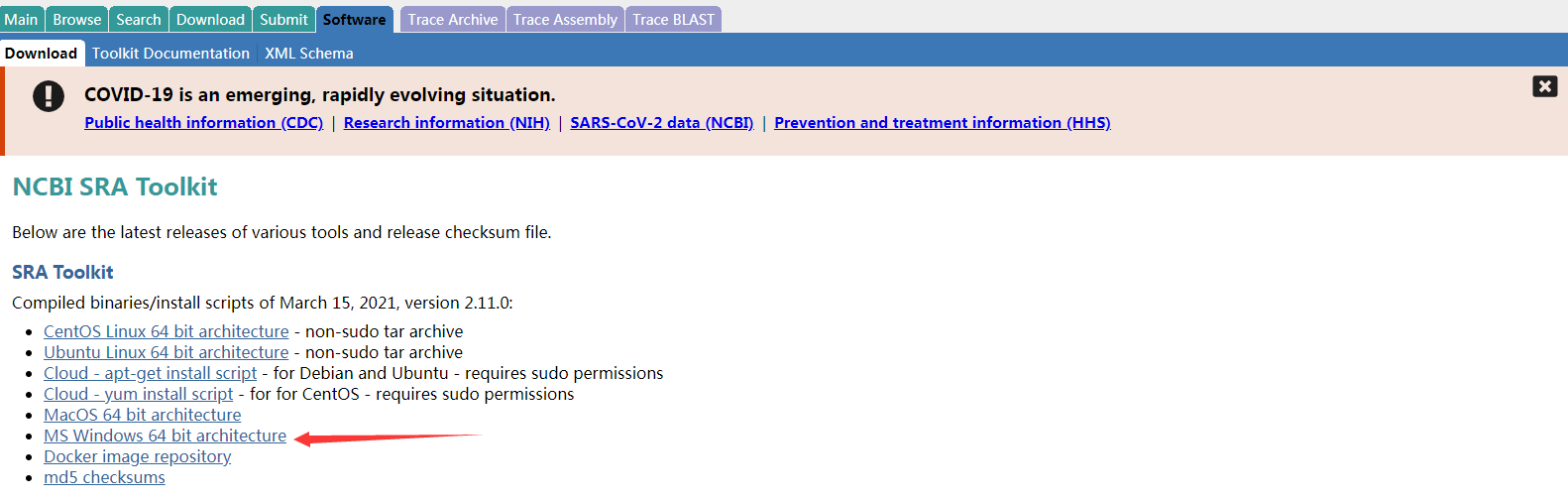
5、根據操作系統選擇相應版本的軟件下載。Window版本軟件下載后為一個壓縮文件 sratoolkit.2.11.0-win64.zip。解壓縮后,設置環境變量后就可以使用。
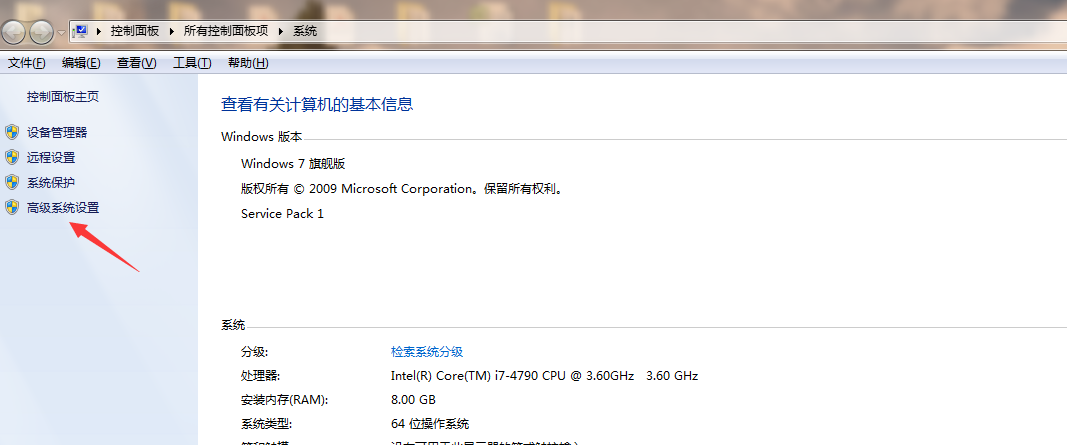
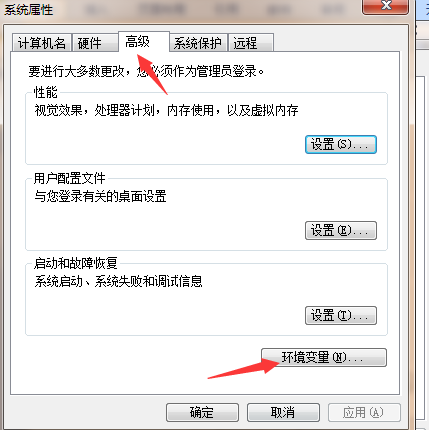
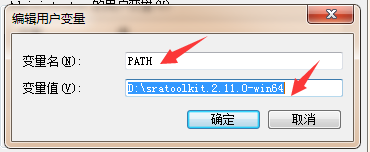
具體安裝方法:鼠標移至“計算機”圖標,點擊右鍵---屬性---高級系統設置---環境變量---PATH---新建---輸入sratoolkit.2.11.0-win64解壓縮所在的路徑,點擊確認就可以使用了。
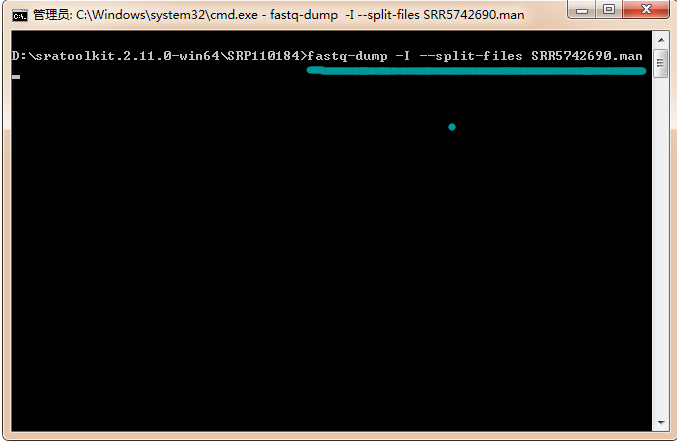
6、SRA下載文件轉化為fastq格式文件 。在測序下載文件中,按shift鍵同時點擊右鍵,在出來的下拉框中點擊“在此處打開命令框”,出現dos界面。輸入命令fastq-dump -I --split-files SRR5742690.man, SRR5742690.man為下載數據文件名及后綴。程序運行完成后會兩個fastq文件,是測序數據的兩個雙端文件。
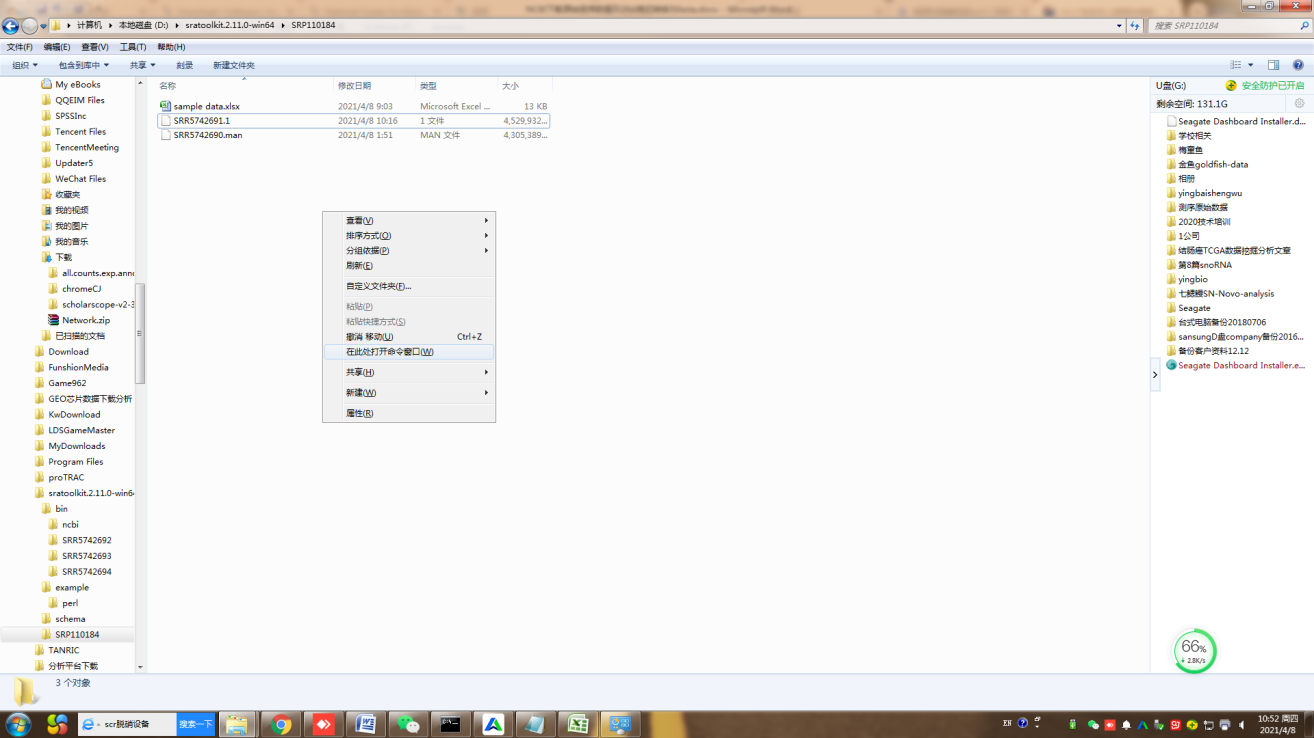

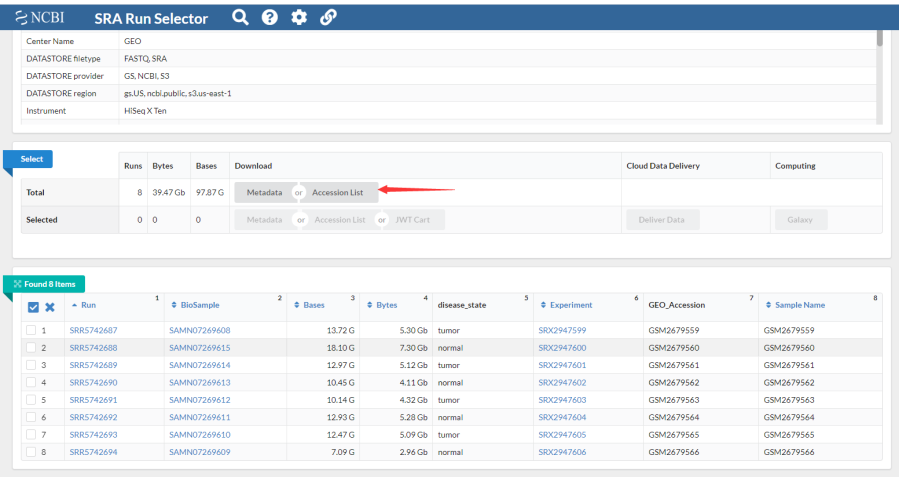
7、如果樣本比較多可以通過NCBI提供的下載工具SRA Toolkit下載。在SRA Run Selector界面下載所有樣本信息Accession list(下載得到SRR_Acc_List.txt文件)。
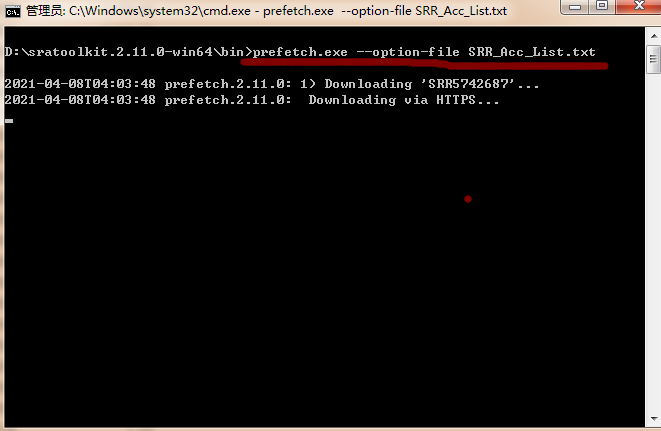
8、將RR_Acc_List.txt放到sratoolkit.2.11.0-win64文件夾中bin文件夾中,在bin文件夾中按住shift同時點擊郵件打開dos命令框,輸入命令:prefetch.exe --option-file SRR_Acc_List.txt。程序會逐個下載各個樣本的數據,一個樣本一個文件夾。
9、得到fastq雙端測序數據之后就可以進行后續差異篩選,功能富集等分析了。